ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 30.07.2024
Просмотров: 30
Скачиваний: 0
8.2. Абсорбционная спектроскопия в
ультрафиолетовой и видимой областях
8.2.1. Общие положения и возможности метода
Абсорбционная спектроскопия в ультрафиолетовой и видимой областях – первый спектральный метод, нашедший широкое применение для исследования органических соединений. Хотя в настоящее время этот вид спектроскопии уступил лидирующее положение другим физическим методам определения строения молекул, его достоинства и сейчас не вызывают сомнений, а в будущем, возможно, даже возрастут как в связи с неуклонным совершенствованием аппаратуры, расширяющим исследуе-мый спектральный диапазон, так и вследствие прогресса в теории спектроскопии.
Спектры поглощения в ультрафиолетовой (УФ-спектры) и видимой областях обусловлены энергетическими переходами между электронными состояниями молекулы, в связи с чем их также называют электронными спектрами.
Электронные спектры поглощения, как и другие методы молекулярной спектроскопии, применяются для решения самых разнообразных задач при исследовании строения и свойств органических соединений и их количественном анализе. Это: определение структуры и идентификация веществ, контроль их чистоты, изучение кинетики и механизма химических реакций, количественное определение состава одно- и многокомпонентных систем, измерения констант диссоциации кислот и оснований, исследование процессов комплексообразования и т.д.
Каждое электронное состояние молекулы характеризуется некоторым интервалом значений энергии, связанным с колебательным движением молекулы. Поэтому любому электронному переходу в спектре соответствует широкая полоса поглощения. При съемке спектра в газовой фазе, как правило, удается выявить колебательную структуру электронного перехода (в таком случае полоса поглощения выглядит как система близко расположенных узких полос), но при получении спектра в конденсированной фазе очень часто (но не всегда) тонкая структура полосы полностью исчезает вследствие проявления межмолекулярных взаимодействий.
Электронные спектры (ближняя ультрафиолетовая и видимая области) характеризуют только сопряженные системы кратных связей, ароматические структуры, функциональные группы с гетероатомами и - связями (CO, NO, NO2 и др.). В пределах этих фрагментов, являющихся "хромофорами", метод дает ценную информацию, включая некоторые данные о конфигурации и степени разветвленности скелета. Однако никаких данных о структуре удаленных от хромофорных частей молекулы, предельных соединений и соединений с изолированными двойными или тройными углерод - углеродными связями электронная спектроскопия не дает.
Электронные переходы в молекуле классифицируют в соответствии с типом содержащихся в ней валентных электронов. Электроны, образующие простую связь, носят название -электронов, образующие двойную (тройную) связь – -электронов. Кроме того, в молекулах, содержащих атомы таких элементов как кислород, азот и т.д., существуют неспаренные, или n-электроны.
В соответствии с этим различают -, -, и n-орбитали, которые, как правило, в основном состоянии заняты электронами. При возбуждении молекулы квантом света возможны переходы электронов со связывающей ( ) или несвязывающей (n) орбитали на разрыхляющую орбиталь (* или *) с более высокой энергией, которая в основном состоянии свободна (вакантна). Эти переходы реализуются только в молекулах, содержащих ненасыщенные группировки. Атомную группировку, которая придает соединению способность и избирательному поглощению в ближнем ультрафиолете или видимой области, называют хромофором.
Хромофоры подразделяются на изолированные и сопряженные. К первым относят группировки с одной кратной связью, такие как С = С, С = О, N = N и т.п., а ко вторым – структурные элементы, представляющие собой системы сопряженных кратных связей. Соединение, содержащее сопряженный хромофор, поглощает в более длинноволновой области и с большей интенсивностью, чем соединение, включающее те же, но изолированные хромофоры. В последнем случае спектр полифункциональ-ного соединения можно трактовать просто как результат суммирования поглощения соответствующих изолированных кратных связей. Некоторые из хромофоров (например, сопряженный хромофор С = С – С = С), обеспечивают поглощение в ближнем ультрафиолете за счет только *-перехода, другие (как изолированный хромофор С = О) – за счет *-перехода, а третьи (например, сопряженный хромофор С = С – С = О) – вследствие реализации как *-, так и n *-переходов.
Атомную группировку, не содержащую кратных связей, которая не имеет максимума поглощения в ближнем ультрафиолете, но включение которой в систему хромофора приводит к увеличению длины волны *-перехода и увеличению интенсивности поглощения, называют ауксохромом.
Типичными ауксохромами являются ОН, NH2, SH, т.е. группы, содержащие гетероатом со свободной электронной парой.
При выявлении взаимосвязи спектра со структурой молекулы бывает целесообразным наблюдение за изменениями в положении и интенсив-ности полос поглощения при переходе от некоторого родоначального хромофора, ответственного за поглощение, к модифицированному. Для этого в систему вводят дополнительную хромофорную или ауксохромную группу. Для характеристики спектральных изменений, вызванных модификацией структуры, используется специальная терминология:
батохромный сдвиг – смещение полосы поглощения в сторону более длинных волн;
гипсохромный сдвиг – смещение полосы поглощения в сторону более коротких волн;
гиперхромный эффект – увеличение интенсивности поглощения;
гипохромный эффект – уменьшение интенсивности поглощения.
Эти же термины используются и для описания изменений в спектре, вызываемых заменой растворителя.
8.2.2. Условия получения и способы изображения
электронных спектров
Спектрометры регистрируют УФ-спектр вещества в виде графика зависимости интенсивности поглощения, выраженной в единицах оптической плотности А, от длины волны (или волнового числа /). Типичный УФ-спектр представлен на рис. 8.9.
0

0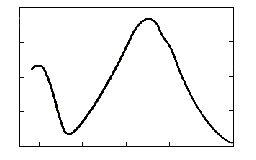
Рис. 8.9. УФ-спектр коричной кислоты
По определению, оптическая плотность А есть десятичный логарифм отношения интенсивностей светового луча до и после прохождения слоя вещества:
А = lgI0/I
В соответствии с законом Бугера – Ламберта – Бера, оптическая плотность раствора поглощающего вещества в прозрачном растворителе пропорциональна концентрации с и толщине слоя (см) раствора. В УФ-спектроскопии принято концентрацию выражать в моль/л. В этом случае
А = l с,где – молярный коэффициент поглощения, его размерность л/(моль . см).
Именно (а не А) является мерой поглощающей способности молекул вещества, независимой от его макроскопических параметров. Поэтому УФ-спектр следует представлять как функцию от длины волны. Очень часто удовлетворяются переводом А в только для максимумов на рабочей кривой А = f () (рис.8.9). Понятно, что рассчитать величину можно, лишь зная молекулярную массу (М) соединения. Если таковая неизвестна, то в качестве интенсивной характеристики поглощающей способности вещества используют коэффициент поглощения а: = аМ.
Полосы поглощения в электронных спектрах органических молекул могут сильно различаться по величине (от 10 до 105). Поскольку рабочая кривая А = f (), имеющая размеры по ординате примерно от 0 до 2, позволяет фиксировать полосы, различающиеся по величине менее чем в 100 раз, то для получения полного многополосного спектра необходимо проводить отдельные измерения для полос, сильно различающихся по интенсивности, используя растворы вещества различной концентрации (рис.8.9а). В этом случае единую спектральную кривую строят вручную на основании отдельных кривых А = f (), переходя по оси ординат на логарифмическую шкалу. Получающаяся кривая lg = f () дает возможность наглядно и с одинаковой точностью представить на одном графике участки спектра, отличающиеся по интенсивности на несколько порядков (рис.8.10 б).
В последнее время в изображении спектров наблюдается тенденция к переходу от шкалы длин волн к шкале волновых чисел /. При такой замене вид спектральной кривой несколько изменяется. В частности, на кривых lg = f (/) полосы поглощения имеют более симметричный контур и располагаются более или менее равномерно вдоль оси абсцисс (рис. 8.2.11).
Ч асто
возникает необходимость охарактеризовать
УФ-спектр, не прибегая к графическому
изображению. В таком случае принято
перечислять координаты максимумов
кривой поглощения. Например, спектр,
представленный на рис.5.1, записывают
следующим образом:
асто
возникает необходимость охарактеризовать
УФ-спектр, не прибегая к графическому
изображению. В таком случае принято
перечислять координаты максимумов
кривой поглощения. Например, спектр,
представленный на рис.5.1, записывают
следующим образом:
![]() ,
нм ():
220 (13900), 272 (21600)
,
нм ():
220 (13900), 272 (21600)
250 300 350 нм А 0,8 0,4 0,2 0,6
250 300 350 нм 2 2 3 1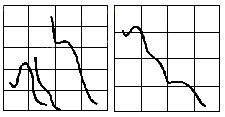

а
б
Рис. 8.10. УФ-спектры фенилбензилкетона в изооктане:
а) спектр в форме А = f ();
б) спектр в форме lg = f ()

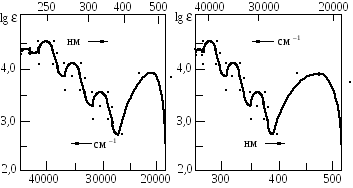
а
б
Рис. 8.11. УФ-спектры 1,4-дигидроантрахинона в метаноле:
а) спектр в форме lg = f (/); б) спектр в форме lg = f ()
Электронные спектры могут быть получены для любого агрегатного состояния вещества. Однако практически используется лишь съемка спектра раствора вещества, т.к. только в этом случае возможно точно измерить интенсивность поглощения излучения при простом способе приготовления образца. Для УФ-области спектра выше 200 нм прозрачны любые растворители. Из неполярных инертных растворителей таковыми являются: гексан, гептан, циклогексан, а из полярных активных (т.е. способных эффективно сольватировать растворенное вещество) – вода, метанол, 95%-ный этанол, ацетонитрил, диоксан. Материал кювет УФ-спектрофото-метров – кварц – позволяет использовать в качестве активного растворителя даже такие агрессивные жидкости, как спиртовые и водные растворы щелочей и минеральных кислот. При замене растворителя УФ-спектр вещества всегда несколько изменяется, поэтому ссылка на используемый растворитель необходима при сообщении данных по спектру вещества.
Во многих случаях для записи УФ-спектра необходимо работать с разбавленными растворами. Так, для соединения с молекулярной массой около 100, имеющего 10000, условию А = 0,5 (оптимальный интервал для измерения оптической плотности на большинстве спектрофотометров находится вблизи А = 0,5 0,2) при = 1 см соответствует концентрация раствора порядка 0,005 г/л. Учитывая, что при использовании сантиметровых кювет необходимый объем раствора не превышает 5 мл, получается, что навеска вещества для этого должна быть порядка 0,025 мг. Поскольку оперировать со столь малыми навесками затруднительно, то обычно на аналитических весах, обеспечивающих определение сотых долей миллиграмма, берется навеска порядка 2-3 мг, которая растворяется в точно фиксированном объеме растворителя, например в 20 мл. Затем, посредством отбора аликвотной части раствора и ее разбавления растворителем, получают раствор требуемой концентрации.
При интерпретации спектров для целей структурного анализа целесообразно различать три вида полос:
1. Очень интенсивные полосы с 103, соответствующие *-переходам, типичные для конъюгированных систем и часто обозначаемые в литературе как К-полосы. Аналогичные по интенсивности полосы *-переходов в ароматических системах обозначаются как Е-полосы (Е1 – соответствует разрешенным по симметрии переходам с 104 - 105, а Е2 – запрещенным переходам с 2000 - 12000).
2. Слабые полосы n *-переходов с 102, характерные для непредельных гетероатомных функциональных групп и радикалов, так называемые R-полосы.
3. Полосы средней интенсивности ( от 102 до 103), соответствующие запрещенным *-переходам в ароматических структурах бензольного типа (В-полосы).
Важным экспериментальным критерием отнесения полосы к *- или n *-переходам является направление смещения максимума поглощения (правило Мак – Конелла) при переходе от неполярных растворителей к полярным. Для n *-переходов в этом случае наблюдается "синий" или гипсохромный (в сторону более коротких волн), а для *-переходов чаще "красный" или батохромный (длинноволновый) сдвиг.
Самым надежным критерием отнесения полосы к переходу с участием несвязывающих электронов (n *) является ее исчезновение в кислых средах. Это объясняется протонированием n‑электронов неподеленной пары, в результате чего они теряют свой несвязывающий характер, и поэтому переход вообще не наблюдается.
В целом следует отметить, что УФ-спектры чаще всего дают лишь ориентировочную информацию о структуре вещества, которая не является достаточной для окончательных выводов по его идентификации.