Файл: Реферат На тему адсорбция Студент Преподаватель Екатеринбург 2006г.rtf
Добавлен: 04.12.2023
Просмотров: 134
Скачиваний: 7
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Мицеллярные структуры 1 и 2 относятся к гидрофильным золям, а 3 и 4 — к органофильным. Сферические М. (1 и 3) при разбавлении системы ниже критической концентрации мицеллообразования обратимо распадаются на отдельные молекулы или димеры (подробнее см. Полуколлоидные системы). При более высоких концентрациях сферические М. превращаются в пластинчатые (2 и 4). Последние, взаимодействуя между собой, способны создавать в объёме системы структурную сетку геля (см. Гели, Дисперсная структура).
Наличием М. объясняется моющее действие водных растворов (точнее, коллоидных дисперсий) мыл, а также некоторые явления в биологических системах и при технологических процессах (см. также Солюбилизация).
Коагуляция (от лат. Coagulatio — свёртывание, сгущение), слипание частиц коллоидной системы при их столкновениях в процессе теплового (броуновского) движения, перемешивания или направленного перемещения во внешнем силовом поле. В результате К. образуются агрегаты — более крупные (вторичные) частицы, состоящие из скопления более мелких (первичных). Первичные частицы в таких скоплениях соединены силами межмолекулярного взаимодействия непосредственно или через прослойку окружающей (дисперсионной) среды. К. сопровождается прогрессирующим укрупнением частиц (увеличением размера и массы агрегатов) и уменьшением их числа в объёме дисперсионной среды — жидкости или газа.
Различают быструю и медленную К. При быстрой К. почти каждое соударение частиц эффективно, т. е. приводит к их соединению; при медленной К. соединяется часть сталкивающихся частиц. В жидкой среде, например при К. золей, укрупнение частиц до известного предела (приблизительно до размера 10-4 см) не сопровождается их оседанием или всплыванием. Это скрытая К., при которой система сохраняет седиментационную устойчивость. Дальнейший рост частиц приводит к образованию сгустков или хлопьев (флокул), выпадающих в осадок (коагулят, коагель) или скапливающихся в виде сливок у поверхности; это явная К. В некоторых случаях при К. во всём объёме дисперсионной среды возникает рыхлая пространственная сетка (коагуляционная структура) и расслоения системы не происходит (см. Гели). Если коллоидные частицы — капельки жидкости или пузырьки газа, то К. может завершиться их слиянием, коалесценцией.
К. — самопроизвольный процесс, который, в соответствии с законами термодинамики
, является следствием стремления системы перейти в состояние с более низкой свободной энергией. Однако такой переход затруднен, а иногда практически невозможен, если система агрегативно устойчива, т. е. способна противостоять укрупнению (агрегированию) частиц. Защитой от К. при этом может быть электрический заряд и (или) адсорбционно-сольватный слой на поверхности частиц, препятствующий их сближению (подробнее см. Коллоидные системы). Нарушить агрегативную устойчивость можно, например, повышением температуры (термокоагуляция), перемешиванием или встряхиванием, введением коагулирующих веществ (коагулянтов) и др. видами внешнего воздействия на систему. Минимальная концентрация введенного вещества, электролита или неэлектролита, вызывающая К. в системе с жидкой дисперсионной средой, называется порогом коагуляции. В полидисперсных системах, где частицы имеют разную величину, можно наблюдать ортокинетическую К. — налипание мелких частиц на более крупные при их оседании или всплывании. Слипание однородных частиц называется гомокоагуляцией, а разнородных — гетерокоагуляцией или адагуляцией. Гетерокоагуляция часто происходит при смешении дисперсных систем различного состава. К. может наступить без какого-либо внешнего воздействия на коллоидную систему (автокоагуляция) как результат физических или химических изменений, происходящих при её старении. Иногда К. обратима; в благоприятных условиях, особенно при введении поверхностно-активных веществ, понижающих поверхностную межфазную энергию и облегчающих диспергирование, возможен распад агрегатов на первичные частицы (пептизация) и переход коагеля в золь.
К. играет важную роль во многих технологических, биологических, атмосферных и геологических процессах. Так, при нагревании биополимеров (белков, нуклеиновых кислот) и при некоторых др. воздействиях на них, например изменении pH, наблюдается их К. Явления К. во многих биологических дисперсных системах (например, крови, лимфе) важны в связи с вопросами их агрегативной устойчивости. Очистка природных и сточных вод от высокодисперсных механических примесей, борьба с загрязнением воздушного пространства аэрозолями,выделение каучука из латекса, получение сливочного масла и др. пищевых продуктов — характерные примеры использования К. в практических целях. Нежелательна К. при получении и хранении суспензий,эмульсий, порошков и др. дисперсных систем промышленного или бытового назначения.
Топохимические реакции, реакции химические, происходящие на границе раздела твёрдых фаз. Примеры Т. р.: дегидратация кристаллогидратов, восстановление окислов, термический распад азидов тяжёлых металлов и т.д. Особенности Т. р.: 1) они начинаются не во всём объёме, а с отдельных, наиболее реакционноспособных мест твёрдого тела (локализация процесса); 2) возникнув в каком-то месте, реакция продолжается в соседних областях кристалла (автолокализация процесса). Причины локализации процесса при Т. р. обычно связаны с наличием дефектов в кристаллах и малой подвижностью ионов, атомов или молекул, образующих кристаллическую решётку. Автолокализация процесса обусловлена каталитическим влиянием твёрдого или газообразного продукта реакции, а также кристаллохимическими особенностями развития реакции в кристалле. Межфазовая поверхность, в пределах которой локализованы Т. р., возникает вследствие образования и роста реакционных ядер; скорость процесса обычно пропорциональна величине этой поверхности в каждый данный момент времени. Поэтому кинетический анализ Т. р. включает не только учёт развития процесса во времени, но и в пространстве. Значит, влияние на скорость Т. р. оказывают дефекты в кристаллах. Оно проявляется в изменении как числа потенциальных центров реакции на поверхности, так и условий для явлений переноса в твёрдом теле. С существенной ролью дефектов в развитии Т. р. связаны также широко известный эффект влияния «предыстории» препарата (реагента) на его реакционную способность, многообразие факторов, воздействующих на их скорость, и т.д. Характер влияния дефектов в кристаллах на скорости Т. р. в каждом конкретном случае зависит как от вида и концентрации дефектов, так и от механизма элементарных стадий.
Т. р. широко используются на практике. К числу наиболее важных Т. р. относятся процессы обжига, восстановления, хлорирования руд многих металлов, цементация стали, производство керамики и огнеупоров, приготовление катализаторов, получение ферритов, некоторые стадии фотографического процесса, газовая коррозия металлов и сплавов. Во многих случаях разложение взрывчатых веществ при нагревании, процессы синтеза и очистки полупроводниковых материалов также относятся к Т. р.
Катализ (от греч. katálysis — разрушение), изменение скорости химических реакций в присутствии веществ (
катализаторов),вступающих в промежуточное химическое взаимодействие с реагирующими веществами, но восстанавливающих после каждого цикла промежуточных взаимодействий свой химический состав. Реакции с участием катализаторов называются каталитическими. Количество реагирующего вещества, которое может испытать превращение в присутствии определённого количества катализатора, не ограничивается какими-либо стехиометрическими соотношениями и может быть очень большим. Этим каталитические реакции отличаются от индуцируемых, или сопряжённых реакций, когда одна реакция вызывается или ускоряется (индуцируется) другой и происходит необратимое превращение вещества-индуктора. Возможные изменения катализатора при каталитических реакциях являются результатом побочных процессов, ни в коей мере не обусловливающих каталитическое действие.
Воздействие катализатора открывает новый реакционный путь, обычно с большим числом стадий, на котором катализатор входит в состав активного комплекса (активированного комплекса) по крайней мере одной из стадий. Если при этом скорость реакции становится больше, чем в отсутствие катализатора, то К. называется положительным (его нередко отождествляют с общим понятием К.). Возможен и обратный случай, когда происходит отрицательный К.: в присутствии катализатора исключается один из возможных путей реакции и остаются лишь более медленные, в результате чего реакция замедляется или даже практически полностью подавляется (см. Антиокислители, Ингибиторы химические). Особый случай К. — ускорение реакции при воздействии продукта реакции или одного из промежуточных веществ, образующихся при реакции (см. Автокатализ).
К. не связан с изменением свободной энергии катализатора, и воздействие катализатора не может поэтому смещать положение равновесия химической реакции. Вблизи состояния равновесия катализаторы в равной степени ускоряют как прямую, так и обратную реакцию.
Основным фактором, определяющим скорость химического превращения, является энергия активации (Е) — разность энергий активного комплекса и исходных реагирующих молекул. Если предположить, что реакция не нарушает равновесного распределения энергии между молекулами, то вероятность образования активного комплекса, а следовательно, и скорость реакции в первом приближении пропорциональна exp (—E/RT), где R — газовая постоянная
, Т — абсолютная температура. Отсюда следует, что скорость реакции тем больше, чем меньше Е, и вследствие экспоненциальной зависимости возрастает значительно даже при небольшом снижении Е.
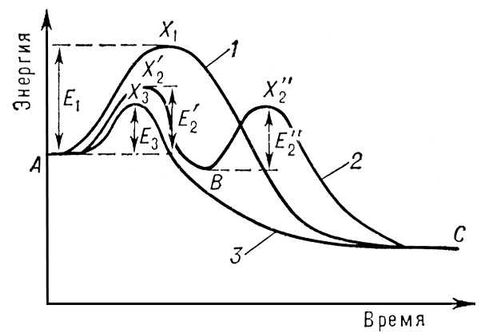
На рис. представлено изменение энергии при реакции без катализатора (кривая 1) и при участии катализатора (кривые 2 и 3). Кривая 2 с двумя максимумами соответствует образованию одного промежуточного продукта. Число стадий и промежуточных продуктов часто бывает значительно большим. Взаимодействие реагирующих веществ с катализатором может и не приводить к образованию стабильной формы промежуточного соединения (кривая 3). Но и в этом случае катализатор входит в состав активного комплекса и взаимодействие реагирующих веществ с катализатором определяет реакционный путь. Если энергии активных комплексов всех стадий реакционного пути с участием катализатора ниже энергии активного комплекса реакции без катализатора (т. е. , и E3 ниже E1), то участие катализатора приведёт к увеличению скорости (положительный К.). Во многих случаях К. ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции). Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный К. часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.
, и E3 ниже E1), то участие катализатора приведёт к увеличению скорости (положительный К.). Во многих случаях К. ускорение реакции достигается благодаря появлению богатых энергией частиц в процессе самой реакции, причём их концентрация может превосходить равновесную (см. Цепные реакции). Например, каталитическое воздействие воды на окисление окиси углерода связано с развитием реакционных путей с участием гидроксильных групп и атомов водорода. Отрицательный К. часто связан с прекращением цепной реакции вследствие обрыва цепей при взаимодействии отрицательного катализатора с активными частицами. Примером может служить замедляющее влияние кислорода на соединение водорода с хлором.
Характер промежуточного химического взаимодействия при К. весьма разнообразен. Обычно различают две группы каталитических процессов: кислотно-основной (гетеролитический) и окислительно-восстановительный (гомолитический). В процессах первой группы происходит промежуточное кислотно-основное взаимодействие реагирующих веществ с катализатором, например переход протона от катализатора к реагирующим веществам или наоборот. На последующих стадиях протон перемещается в обратном направлении, и катализатор восстанавливает свой состав. При К. апротонными кислотами взаимодействие осуществляется через свободную пару электронов реагирующего вещества. Примерами кислотно-основного К. могут служить гидролиз сложных эфиров, ускоряемый кислотами; гидратация олефинов в присутствии фосфорно-кислотных катализаторов; изомеризация и крекинг углеводородов на алюмосиликатных катализаторах; алкилирование; полимеризация и многие другие реакции. При реакциях окислительно-восстановительного К. промежуточное взаимодействие связано с электронными переходами между катализатором и реагирующими веществами. К этой группе относятся окисление двуокиси серы в трёхокись в производстве серной кислоты; окисление аммиака до окиси азота при получении азотной кислоты; многочисленные процессы парциального окисления органических соединений, например этилена в окись этилена, нафталина во фталевый ангидрид; гидрогенизация; дегидрогенизация; циклизация и ароматизация углеводородов; разложение перекиси водорода и многие др. Каталитической активностью в отношении окислительно-восстановительных реакций обладают преимущественно металлы 4-, 5- и 6-го периодов системы Д. И. Менделеева, имеющие недостроенную
Наличием М. объясняется моющее действие водных растворов (точнее, коллоидных дисперсий) мыл, а также некоторые явления в биологических системах и при технологических процессах (см. также Солюбилизация).
Коагуляция (от лат. Coagulatio — свёртывание, сгущение), слипание частиц коллоидной системы при их столкновениях в процессе теплового (броуновского) движения, перемешивания или направленного перемещения во внешнем силовом поле. В результате К. образуются агрегаты — более крупные (вторичные) частицы, состоящие из скопления более мелких (первичных). Первичные частицы в таких скоплениях соединены силами межмолекулярного взаимодействия непосредственно или через прослойку окружающей (дисперсионной) среды. К. сопровождается прогрессирующим укрупнением частиц (увеличением размера и массы агрегатов) и уменьшением их числа в объёме дисперсионной среды — жидкости или газа.
Различают быструю и медленную К. При быстрой К. почти каждое соударение частиц эффективно, т. е. приводит к их соединению; при медленной К. соединяется часть сталкивающихся частиц. В жидкой среде, например при К. золей, укрупнение частиц до известного предела (приблизительно до размера 10-4 см) не сопровождается их оседанием или всплыванием. Это скрытая К., при которой система сохраняет седиментационную устойчивость. Дальнейший рост частиц приводит к образованию сгустков или хлопьев (флокул), выпадающих в осадок (коагулят, коагель) или скапливающихся в виде сливок у поверхности; это явная К. В некоторых случаях при К. во всём объёме дисперсионной среды возникает рыхлая пространственная сетка (коагуляционная структура) и расслоения системы не происходит (см. Гели). Если коллоидные частицы — капельки жидкости или пузырьки газа, то К. может завершиться их слиянием, коалесценцией.
К. — самопроизвольный процесс, который, в соответствии с законами термодинамики
, является следствием стремления системы перейти в состояние с более низкой свободной энергией. Однако такой переход затруднен, а иногда практически невозможен, если система агрегативно устойчива, т. е. способна противостоять укрупнению (агрегированию) частиц. Защитой от К. при этом может быть электрический заряд и (или) адсорбционно-сольватный слой на поверхности частиц, препятствующий их сближению (подробнее см. Коллоидные системы). Нарушить агрегативную устойчивость можно, например, повышением температуры (термокоагуляция), перемешиванием или встряхиванием, введением коагулирующих веществ (коагулянтов) и др. видами внешнего воздействия на систему. Минимальная концентрация введенного вещества, электролита или неэлектролита, вызывающая К. в системе с жидкой дисперсионной средой, называется порогом коагуляции. В полидисперсных системах, где частицы имеют разную величину, можно наблюдать ортокинетическую К. — налипание мелких частиц на более крупные при их оседании или всплывании. Слипание однородных частиц называется гомокоагуляцией, а разнородных — гетерокоагуляцией или адагуляцией. Гетерокоагуляция часто происходит при смешении дисперсных систем различного состава. К. может наступить без какого-либо внешнего воздействия на коллоидную систему (автокоагуляция) как результат физических или химических изменений, происходящих при её старении. Иногда К. обратима; в благоприятных условиях, особенно при введении поверхностно-активных веществ, понижающих поверхностную межфазную энергию и облегчающих диспергирование, возможен распад агрегатов на первичные частицы (пептизация) и переход коагеля в золь.
К. играет важную роль во многих технологических, биологических, атмосферных и геологических процессах. Так, при нагревании биополимеров (белков, нуклеиновых кислот) и при некоторых др. воздействиях на них, например изменении pH, наблюдается их К. Явления К. во многих биологических дисперсных системах (например, крови, лимфе) важны в связи с вопросами их агрегативной устойчивости. Очистка природных и сточных вод от высокодисперсных механических примесей, борьба с загрязнением воздушного пространства аэрозолями,выделение каучука из латекса, получение сливочного масла и др. пищевых продуктов — характерные примеры использования К. в практических целях. Нежелательна К. при получении и хранении суспензий,эмульсий, порошков и др. дисперсных систем промышленного или бытового назначения.
Топохимические реакции, реакции химические, происходящие на границе раздела твёрдых фаз. Примеры Т. р.: дегидратация кристаллогидратов, восстановление окислов, термический распад азидов тяжёлых металлов и т.д. Особенности Т. р.: 1) они начинаются не во всём объёме, а с отдельных, наиболее реакционноспособных мест твёрдого тела (локализация процесса); 2) возникнув в каком-то месте, реакция продолжается в соседних областях кристалла (автолокализация процесса). Причины локализации процесса при Т. р. обычно связаны с наличием дефектов в кристаллах и малой подвижностью ионов, атомов или молекул, образующих кристаллическую решётку. Автолокализация процесса обусловлена каталитическим влиянием твёрдого или газообразного продукта реакции, а также кристаллохимическими особенностями развития реакции в кристалле. Межфазовая поверхность, в пределах которой локализованы Т. р., возникает вследствие образования и роста реакционных ядер; скорость процесса обычно пропорциональна величине этой поверхности в каждый данный момент времени. Поэтому кинетический анализ Т. р. включает не только учёт развития процесса во времени, но и в пространстве. Значит, влияние на скорость Т. р. оказывают дефекты в кристаллах. Оно проявляется в изменении как числа потенциальных центров реакции на поверхности, так и условий для явлений переноса в твёрдом теле. С существенной ролью дефектов в развитии Т. р. связаны также широко известный эффект влияния «предыстории» препарата (реагента) на его реакционную способность, многообразие факторов, воздействующих на их скорость, и т.д. Характер влияния дефектов в кристаллах на скорости Т. р. в каждом конкретном случае зависит как от вида и концентрации дефектов, так и от механизма элементарных стадий.
Т. р. широко используются на практике. К числу наиболее важных Т. р. относятся процессы обжига, восстановления, хлорирования руд многих металлов, цементация стали, производство керамики и огнеупоров, приготовление катализаторов, получение ферритов, некоторые стадии фотографического процесса, газовая коррозия металлов и сплавов. Во многих случаях разложение взрывчатых веществ при нагревании, процессы синтеза и очистки полупроводниковых материалов также относятся к Т. р.
Катализ (от греч. katálysis — разрушение), изменение скорости химических реакций в присутствии веществ (
катализаторов),вступающих в промежуточное химическое взаимодействие с реагирующими веществами, но восстанавливающих после каждого цикла промежуточных взаимодействий свой химический состав. Реакции с участием катализаторов называются каталитическими. Количество реагирующего вещества, которое может испытать превращение в присутствии определённого количества катализатора, не ограничивается какими-либо стехиометрическими соотношениями и может быть очень большим. Этим каталитические реакции отличаются от индуцируемых, или сопряжённых реакций, когда одна реакция вызывается или ускоряется (индуцируется) другой и происходит необратимое превращение вещества-индуктора. Возможные изменения катализатора при каталитических реакциях являются результатом побочных процессов, ни в коей мере не обусловливающих каталитическое действие.
Воздействие катализатора открывает новый реакционный путь, обычно с большим числом стадий, на котором катализатор входит в состав активного комплекса (активированного комплекса) по крайней мере одной из стадий. Если при этом скорость реакции становится больше, чем в отсутствие катализатора, то К. называется положительным (его нередко отождествляют с общим понятием К.). Возможен и обратный случай, когда происходит отрицательный К.: в присутствии катализатора исключается один из возможных путей реакции и остаются лишь более медленные, в результате чего реакция замедляется или даже практически полностью подавляется (см. Антиокислители, Ингибиторы химические). Особый случай К. — ускорение реакции при воздействии продукта реакции или одного из промежуточных веществ, образующихся при реакции (см. Автокатализ).
К. не связан с изменением свободной энергии катализатора, и воздействие катализатора не может поэтому смещать положение равновесия химической реакции. Вблизи состояния равновесия катализаторы в равной степени ускоряют как прямую, так и обратную реакцию.
Основным фактором, определяющим скорость химического превращения, является энергия активации (Е) — разность энергий активного комплекса и исходных реагирующих молекул. Если предположить, что реакция не нарушает равновесного распределения энергии между молекулами, то вероятность образования активного комплекса, а следовательно, и скорость реакции в первом приближении пропорциональна exp (—E/RT), где R — газовая постоянная
, Т — абсолютная температура. Отсюда следует, что скорость реакции тем больше, чем меньше Е, и вследствие экспоненциальной зависимости возрастает значительно даже при небольшом снижении Е.
На рис. представлено изменение энергии при реакции без катализатора (кривая 1) и при участии катализатора (кривые 2 и 3). Кривая 2 с двумя максимумами соответствует образованию одного промежуточного продукта. Число стадий и промежуточных продуктов часто бывает значительно большим. Взаимодействие реагирующих веществ с катализатором может и не приводить к образованию стабильной формы промежуточного соединения (кривая 3). Но и в этом случае катализатор входит в состав активного комплекса и взаимодействие реагирующих веществ с катализатором определяет реакционный путь. Если энергии активных комплексов всех стадий реакционного пути с участием катализатора ниже энергии активного комплекса реакции без катализатора (т. е.
Характер промежуточного химического взаимодействия при К. весьма разнообразен. Обычно различают две группы каталитических процессов: кислотно-основной (гетеролитический) и окислительно-восстановительный (гомолитический). В процессах первой группы происходит промежуточное кислотно-основное взаимодействие реагирующих веществ с катализатором, например переход протона от катализатора к реагирующим веществам или наоборот. На последующих стадиях протон перемещается в обратном направлении, и катализатор восстанавливает свой состав. При К. апротонными кислотами взаимодействие осуществляется через свободную пару электронов реагирующего вещества. Примерами кислотно-основного К. могут служить гидролиз сложных эфиров, ускоряемый кислотами; гидратация олефинов в присутствии фосфорно-кислотных катализаторов; изомеризация и крекинг углеводородов на алюмосиликатных катализаторах; алкилирование; полимеризация и многие другие реакции. При реакциях окислительно-восстановительного К. промежуточное взаимодействие связано с электронными переходами между катализатором и реагирующими веществами. К этой группе относятся окисление двуокиси серы в трёхокись в производстве серной кислоты; окисление аммиака до окиси азота при получении азотной кислоты; многочисленные процессы парциального окисления органических соединений, например этилена в окись этилена, нафталина во фталевый ангидрид; гидрогенизация; дегидрогенизация; циклизация и ароматизация углеводородов; разложение перекиси водорода и многие др. Каталитической активностью в отношении окислительно-восстановительных реакций обладают преимущественно металлы 4-, 5- и 6-го периодов системы Д. И. Менделеева, имеющие недостроенную