ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 11.12.2023
Просмотров: 31
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Дифференциальный диагноз проводят с целиакией, другими бронхолегочными заболеваниями. Лечение симптоматическое: антибиотико-, ферменто-, витамино-, кинези-, диетотерапия, муколитическая и бронхолитическая терапия. Профилактика возможна путем скрининга новорожденных на муковисцидоз, пренатальной диагностики с помощью молекулярно-генетического анализа.
Несовершенный остеогенез.
Несовершенный остеогенез — это гетерогенная группа наследственных заболеваний соединительной ткани, характеризующиеся повышенной ломкостью костей. Суммарная частота составляет 1:20 000. Типы наследования: аутосомно-доминантный (I и IV типы) и аутосомно-рецессивный (II и III типы).
Заболевание обусловлено мутациями структурных генов COL1A1 и COL1A2 а-цепей коллагенов (гены картированы для а, (I) цени — на 17q21-q22, для а2 (I) картирован на 7q21-q22). Мутации в генах а-цепей коллагена первого типа приводят к нарушению его свойств и структуры тканей, содержащих этот коллаген.
Основными клиническими признаками заболевания являются переломы длинных трубчатых костей, реже ребер и ключиц, голубые склеры, снижение слуха. Описаны переломы фаланг пальцев и других костей. Для несовершенного остеогенеза характерны опалесцирующие («янтарные») зубы, гипермобильность суставов, пролапс митрального клапана, деформация грудной клетки, кифосколиоз, остеопороз, грыжи.
Наличие у больного переломов костей, возникших внутриутробно, а также переломов костей при рождении, особенно в сочетании с голубыми склерами, позволяет заподозрить несовершенный остеогенез. Снижение слуха редко развивается ранее 10-летнего возраста.
Пенетрантность заболевания неполная. Пораженные индивиды могут иметь только один из основных клинических признаков заболевания. В данном случае только тщательно проведенный клинико генеалогический анализ помогает поставить правильный диагноз.
Диагноз основан на совокупности клинической картины и клинико-генеалогического анализа. Дифференциальный диагноз проводится с другими скелетными дисплазиями, сопровождающимися деминерализацией костей.
Ахондроплазия.
Ахондроплазия — наследственное заболевание костной системы, характеризующееся ризомелической формой карликовости. Частота заболевания 1:100 000 новорожденных. Тип наследования: аутосомно-доминантный. Более чем 70 % случаев представлено свежими мутациями. Заболевание обусловлено точечными мутациями в гене коллагена COL2A1 (ген картирован на 12ql3-ql4). Ведущими клиническими признаками заболевания являются диспропорциональная карликовость, укорочение проксимальных отделов конечностей, макроцефалия, запавшая переносица,
выраженный поясничный лордоз, изменения костей таза, изменения позвоночника (сужение расстояния между корнями дужек поясничных позвонков, нарастающих в каудальном направлении). Характерны широкие кисти, пальцы в виде трезубца, изодактилия.
Средний рост при рождении составляет 47 см, а средний рост для взрослых составляет 130 см для мужчин и 123 см для женщин. Дети, как правило, отстают в моторном развитии, интеллект нормальный. Встречаются гидроцефалия, описаны частые отиты.
Диагноз ставится на основании характерной клинической картины. Дифференциальный диагноз проводится с различными типами ахондрогенеза.
Синдром Холта—Орама (синдром руки-сердца)
Основными клиническими симптомами являются ВПР верхних конечностей (чаще поражается левая рука): гипоплазия, отсутствие, удвоение, дигитализация I пальца кисти, трехфаланговый I палец. Характерны изменения других пальцев (клинодактилии, синдактилии), гипоплазии лучевой, локтевой, плечевой костей. Описаны аномалии ключиц, лопаток, различные деформации грудной клетки, кифоз, сколиоз. В 85% случаев выявляют пороки сердца: дефекты межпредсердной и межжелудочковой перегородок, открытый артериальный проток, коарктация аорты. В остальных случаях при отсутствии структурного дефекта выявляют аномалии ЭКГ.
Диагноз ставится на основании специфической клинической картины. Дифференциальный диагноз проводится с синдромом панцитопении Фанкони, тромбоцитопении с отсутствием лучевой кости, дефектами лучевой кости, VACTERL-ассоциацией, другими типами синдромов рука-сердце. Наиболее часто встречающимися моногенными заболеваниями из группы факоматозов являются нейрофиброматоз I типа (болезнь Реклингхаузена) и туберозный склероз. Клиническими симптомами, характерными для этой группы заболеваний, являются пигментные пятна и склонность к образованию опухолей.
Нейрофиброматоз I типа (болезнь Реклингхаузена)
Нейрофиброматоз I типа является самым частым наследственным заболеванием из группы факоматозов, характеризующимся предрасположенностью к возникновению опухолей периневрия. Встречается с частотой 1:4000 новорожденных. Тип наследования: аутосомно-доминантный. Ген нейрофиброматоза I типа расположен на длинном плече 17-й хромосомы. Описано более 100 типов мутаций.
Наиболее частым признаком НФ I являются множественные светло-коричневые пятна типа «кофе с молоком». У 60% больных отдельные гиперпигментные пятна являются врожденными. С возрастом наблюдается тенденция к увеличению их числа. У детей в допубертатный период должно выявляться не менее 5 пятен с диаметром более 5 мм.
Нейрофибромы являются диагностически важным признаком НФ I и могут появляться лишь в позднем детском возрасте или в юности. Опухолевые образования можно обнаружить по ходу нервных стволов, они не связаны с окружающими тканями, плотные на ощупь, 1-2 см в диаметре, безболезненные при пальпации.
Другие диагностические признаки включают: пигментные пятна типа «веснушек» в кожных складках, узелки Лиша на радужной оболочке глаз, глиомы зрительного нерва, специфические костные дисплазии.
Клинические проявления зависят от возраста больного, и поэтому для подтверждения диагноза часто необходимо наблюдать пациента на протяжении нескольких лет. Как правило, на первом году жизни у больных отмечаются только гиперпигментные пятна. Наличие у ребенка пораженного родственника первой степени родства позволяет поставить точный диагноз НФ I уже в этом возрасте. Течение заболевания прогрессирующее. Наиболее опасными проявлениями болезни Реклингхаузена являются опухоли, иногда из-за их злокачественности, иногда из-за места их расположения (черепно-мозговые нервы, малый таз, ЖКТ).
Диагноз основан на совокупности клинических признаков, данных магнитно-резонансной томографии и молекулярно-генетического исследования. Дифференциальный диагноз проводится с другими заболеваниями из группы факоматозов.
-
Многофакторные заболевания - докладчик Алена
Многофакторные заболевания (мультифакториальные заболевания или болезни с наследственной предрасположенностью) - большая группа болезней (более 90%), развитие которых определяется взаимодействием определенных наследственных факторов (мутаций или сочетаний нормальных аллелей разных генов) и факторов среды.
Болезни с наследственной предрасположенностью возникают у лиц с определенным генотипом (сочетание «предрасполагающих» аллелей) при провоцирующем действии факторов внешней среды. Наследственная предрасположенность к заболеванию может иметь моногенную или полигенную природу.
Генетической основой моногенных форм наследственной предрасположенности являются мутации единичных генов, которые, как правило, наследуются по аутосомно-рецессивному (например, недостаточность лактазы) или Х-сцепленному рецессивному типу (например, недостаточность глюкозо-6-фосфатдегидрогеназы). Однако строго соответствующего законам Менделя распределения пораженных потомков в поколениях наблюдаться не будет. Это обусловлено тем, что для проявления данного патологического признака носитель мутации должен обязательно вступить в контакт со специфическим провоцирующим внешним фактором.
В отсутствии подобного контакта мутантный ген остается «молчащим» и не проявляется в виде патологического признака.
Определенными условиями среды для клинического проявления моногенных форм могут быть загрязнители воздушного бассейна, продукты питания, химический состав питьевой воды, различные ксенобиотики (лекарственные препараты) и т. д.
Этиологической основой полигенных заболеваний с наследственной предрасположенностью является взаимодействие аллелей нескольких генов с комплексом внешнесредовых факторов. Соотношение влияния генетических и средовых факторов различно не только для данной болезни, каждого конкретного больного.
При мультифакториальных заболеваниях, т.е. заболеваниях с наследственным предрасположением, основой оценки риска являются эмпирические данные о популяционной и семейной частоте каждого из них.
Специфический генетический риск до 5% принято считать низким, до 10% —повышенным в легкой степени, до 20% —средним, выше 20% — высоким. Генетический риск средней степени расценивают как противопоказание к зачатию или показание к прерыванию уже имеющейся беременности. Возможность проведения пренатальной диагностики является определяющей для принятия положительного решения в отношении завершения беременности.
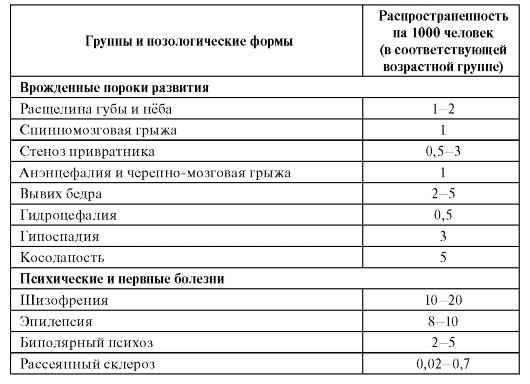
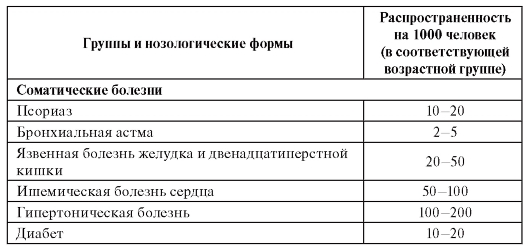
Частота широко распространенных мультифакториальных заболеваний: гипертоническая болезнь (10-20%), ИБС (5-10%), алкоголизм (1,4 - 10%), язвенная болезнь желудка и 12-перстной кишки (2-5%), сахарный диабет и шизофрения (1-2%).
Примеры заболеваний
Полигенная форма
Гипертоническая болезнь - мультифакторное полиэтиологическое заболевание, характеризующееся повышением АД выше 140/90 мм рт.ст., с развитием симптомов поражения сердца, головного мозга, почек, сосудов при условии исключения вторичных артериальных гипертензий.
Подверженность представлена:
1. Вклад генетических факторов – 69%
2. Материнским эффектом – 6%
3. Влияние среды – 25%
Ишемическая болезнь сердца (ИБС) – это заболевание сердечно-сосудистой системы, связанное с нарушением равновесия между объемом коронарного кровообращения и метаболическими потребностями миокарда.
Подверженность представлена:
1. Вклад генетических факторов – 52%
2. Влияние среды – 48%
Язвенная болезнь желудка и ДПК - это мультифакториальное хроническое заболевание, сопровождающееся образованием язв в желудке с возможным прогрессированием и развитием осложнений.
Подверженность представлена:
1. Вклад генетических факторов – 46%
2. Влияние среды – 54%
3. Материнский эффект – 0%
Моногенная форма
Наследственная непереносимость лактозы клинически проявляется повышением газообразования после приема молока или молочных продуктов.
Частота встречаемости лактазной недостаточности сильно зависит от популяции. Частота в популяции: у европейцев - до 20%, у индейцев Америки - 70 -100%.
В кишечнике взрослых гомозигот отсутствует b-галактозидаза, молочный сахар не расщепляется и не всасывается, брожение молочного сахара под действием кишечной флоры ведет к повышенному газобразованию.
Недостаточность глюкозо-6-фосфатдегидрогеназы - распространенное наследственное заболевание, вызываемое дефектом фермента глюкозо-6-фосфатдегидрогеназы, который вырабатывает NADPH и защищает эритроциты от окислительного стресса.
Спровоцировать могут лекарственные средства через 2-3 дня после приема: противомалярийные препараты, сульаниламиды, анальгетики, некоторые химиопрепараты, витамин К, растительные продукты (бобовые, стручковые).
Частота встречаемости:
в областях, эндемичных по малярии, имеет распространенность от 5-25%;
в неэндемичных областях распространенность менее 0,5%.