Файл: 11 Общие сведения о комплексных соединениях 11 Состав комплексных соединений.docx
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 11.01.2024
Просмотров: 79
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Используя теорию КП, объясните природу тетрагонального искажения оксида Mn3O4 (структурный тип шпинели).
11.16. Объясните, используя теорию КП плоскоквадратную геометрию комплекса [Ni(CN)4]2–.
Две атомные орбитали при взаимодействии создают две молекулярные орбитали — связывающую (более низкую по энергии, стабилизированную) и разрыхляющую13 (более высокую по энергии, дестабилизированную).
В связи с этим энергетические уровни, которыми оперирует теория КП и которые в рамках этой теории остаются атомными (т. е. несвязывающими), достаточно условны и имеют смысл только для соединений с высокой степенью ионности связей. Связывающие и разрыхляющие молекулярные орбитали учитываются в теории поля лигандов(ПЛ)14. В отличие от теории
КП, в теории ПЛ рассматриваются волновые функции комплексного иона в целом, а не только комплексообразователя, при этом также учитывается межэлектронное отталкивание d n-конфигураций.
Типичная схема молекулярных орбиталей для октаэдра ML6, в котором лиганды связаны с комплексообразователем только σ-связями, представлена на рис. 11.14: слева и справа от центра показаны энергетические уровни центрального атома (или иона) и группы лигандов, а в центре — уровни комплекса, получающиеся при их комбинировании. При взаимодействии шести орбиталей лигандов с двумя d-орбиталями ( dx2–y2 и dz2 ),

Рис. 11.14. Схема МО для октаэдра [ML6], в котором лиганды связаны с центральным атомом только σ-связями
одной s- и тремя p-орбиталями центрального атома образуются шесть связывающих σ-МО и шесть разрыхляющих σ*-МО. Оставшиеся три d-орбитали (dxy, dyz и dxz) несвязывающие. Разность энергий несвязывающих молекулярных орбиталей π(t2g) иразрыхляющих молекулярных орбиталейσd*(eg) соответствует параметру 10Dq(Δо)в теории КП.Один из примеров такого комплекса ML6 — катион гексаамминкобальта(III) (рис. 11.15); при заселении его молекулярных орбиталей 12 электронов находятся на связывающих σs-, σp- и σd-МО, шесть — на несвязывающих π(t2g)-МО. Выводы о магнитных свойствах и окраске комплекса, которые можно сделать на основании приведенной схемы МО, в целом совпадают с выводами теории ПЛ.
Несвязывающие t2
g-орбитали могут участвовать в π-взаимодействии с соответствующими орбиталями лиганда. Если комплекс содержит лиганды слабого поля (например, галогенид-ионы), энергия t2g-орбиталей которых ниже энергии молекулярных t2g-орбиталей, величина 10Dq(Δо) уменьшается (рис. 11.16, а). В случае лигандов сильного поля (например, СО, NO, CN–), t2g-орбитали которых расположены по энергии выше t2gорбиталей комплекса, величина 10Dq(Δо) увеличивается (рис. 11.16, б). В таких комплексах происходит перенос электронной плотности не только в прямом направлении (от лиганда к центральному атому), но и в обратном — от центрального атома к лиганду. Это дополнительное взаимодействие называют дативной π-связью.

Рис. 11.15. Схема МО для катиона гексаамминкобальта(III) [Co(NH3)6]3

Рис. 11.16. Зависимость параметра 10Dq от энергии π-орбиталей лиганда: а — энергия орбиталей лиганда ниже, чем у t2g-орбиталей металла;
б — энергия орбиталей лиганда выше, чем у t2g-орбиталей металла
Основные эмпирические параметры, которыми оперирует теория ПЛ, — параметр расщепления Dq и параметры Рака=15, характеризующие меж электронное отталкивание. Все эти параметры закономерно изменяются при варьировании природы центрального иона или лигандов; их абсолютные и относительные числовые значения хорошо описывают химию классических комплексных соединений.
При рассмотрении комплексов с позиций теории ПЛ большое значение имеет ковалентный вклад в связь металл—лиганд. При переходе от теории КП к теории ПЛ важное значение имеет параметр Полинга α, который характеризует степень смешения орбиталей металла и лиганда. Если электрон поровну принадлежит орбиталям обоих атомов связи (чисто ковалентный случай, когда электронная пара располагается строго посередине между атомами на линии связи), α = ±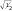 . Другой крайний случай — чисто ионный, при котором взаимодействуют «точечные» заряды (именно его описывает теория КП); при этом α = 1. На практике оба этих крайних варианта встречаются редко.
. Другой крайний случай — чисто ионный, при котором взаимодействуют «точечные» заряды (именно его описывает теория КП); при этом α = 1. На практике оба этих крайних варианта встречаются редко.
Cтепень ионности (ковалентности) можно оценить по так называемому «нефелоксетическому эффекту» — увеличению объема d-орбиталей комплексообразователя из-за их экранирования неподеленными электронными парами лигандов. При этом межэлектронное отталкивание в комплексах оказываются меньшим, чем в соответствующих изолированных ионах металла. Усиление «нефелоксетизма» приблизительно соответствует возрастанию ковалентности связи лиганд—металл; наименьший нефелоксетический эффект оказывает лиганд F–; значительный нефелоксетический эффект у Вr– и CN–. Нефелоксетический эффект можно качественно характеризовать следующим рядом лигандов:
F– < О2– < ОН– < Н2О < ur < NH3 < en < С О2 24− < bipy < СО32− < < CN– < NCS– < Cl– < CNS– < acac < Br– < CNO– < N3– < I0 < S2– < I–
Количественно его определяют из соотношения:
β= Bкомплекс / Bион
где β — величина нефелоксетического эффекта, Bкомплекс — параметр Рака[ для комплекса, а Bион— для несвязанного иона.Параметр Рака[ Вкомплекс зависит от природы лигандов, основной фактор — ионный радиус лиганда. Чем больше ионный радиус, тем больше степень радиального смещения d-электронной плотности и тем меньше межэлектронное отталкивание (а значит, больше величина параметра В).
Теория ПЛ позволяет объяснить закономерности расположения лигандов в спектрохимическом ряду: лиганды располагаются в порядке уменьшения их σ- и π-осно[вности, т. е.в конце ряда находятся лиганды, которые проявляют в основном π-акцепторные свойства.
Следует отметить, что теорию ПЛ (наряду с теорией КП) можно использовать для разных целей:
11.17. Частицы [FеF6]4– и [Fе(CN)6]4– — высокоспиновый и низкоспиновый комплексы соответственно. Составьте энергетическую диаграмму образования связей в этих комплексах и опишите их магнитные свойства.
11.18. Параметр расщепления в поле лигандов 10Dq, определенный из спектроскопических данных, для некоторых комплексов переходных металлов имеет следующие значения:
а) расположите лиганды в порядке возрастания способности к расще-
плению d-подуровней;
б) учитывая, что для комплексов Мn(II) и Со(II) энергия спаривания электронов составляет ∼20 000 см–1, определите, какие из перечисленных комплексов относятся к высокоспиновым.
11.19. Укажите возможную причину того, что магнитный момент иона [Мn(CN)6]3– почти в 2 раза меньше магнитного момента иона [МnF6]3–.
1 КИСТЯКОВСКИЙ Владимир Александрович (1865–1952) — российский и советский физикохимик. Работы по теории растворов, химической термодинамике и электрохимии.
2 ВЕРНЕР Альфред (1866–1919) — швейцарский химик. В 1891–1893 гг. предложил координационную теорию и в последующие годы осуществил ряд экспериментов, доказывающих ее правильность; разработал методы синтеза комплексов и определения их состава и строения. Лауреат Нобелевской премии по химии (1913).
11.16. Объясните, используя теорию КП плоскоквадратную геометрию комплекса [Ni(CN)4]2–.
11.4.3. Метод молекулярных орбиталей
Две атомные орбитали при взаимодействии создают две молекулярные орбитали — связывающую (более низкую по энергии, стабилизированную) и разрыхляющую13 (более высокую по энергии, дестабилизированную).
В связи с этим энергетические уровни, которыми оперирует теория КП и которые в рамках этой теории остаются атомными (т. е. несвязывающими), достаточно условны и имеют смысл только для соединений с высокой степенью ионности связей. Связывающие и разрыхляющие молекулярные орбитали учитываются в теории поля лигандов(ПЛ)14. В отличие от теории
КП, в теории ПЛ рассматриваются волновые функции комплексного иона в целом, а не только комплексообразователя, при этом также учитывается межэлектронное отталкивание d n-конфигураций.
Типичная схема молекулярных орбиталей для октаэдра ML6, в котором лиганды связаны с комплексообразователем только σ-связями, представлена на рис. 11.14: слева и справа от центра показаны энергетические уровни центрального атома (или иона) и группы лигандов, а в центре — уровни комплекса, получающиеся при их комбинировании. При взаимодействии шести орбиталей лигандов с двумя d-орбиталями ( dx2–y2 и dz2 ),
Рис. 11.14. Схема МО для октаэдра [ML6], в котором лиганды связаны с центральным атомом только σ-связями
одной s- и тремя p-орбиталями центрального атома образуются шесть связывающих σ-МО и шесть разрыхляющих σ*-МО. Оставшиеся три d-орбитали (dxy, dyz и dxz) несвязывающие. Разность энергий несвязывающих молекулярных орбиталей π(t2g) иразрыхляющих молекулярных орбиталейσd*(eg) соответствует параметру 10Dq(Δо)в теории КП.Один из примеров такого комплекса ML6 — катион гексаамминкобальта(III) (рис. 11.15); при заселении его молекулярных орбиталей 12 электронов находятся на связывающих σs-, σp- и σd-МО, шесть — на несвязывающих π(t2g)-МО. Выводы о магнитных свойствах и окраске комплекса, которые можно сделать на основании приведенной схемы МО, в целом совпадают с выводами теории ПЛ.
Несвязывающие t2
g-орбитали могут участвовать в π-взаимодействии с соответствующими орбиталями лиганда. Если комплекс содержит лиганды слабого поля (например, галогенид-ионы), энергия t2g-орбиталей которых ниже энергии молекулярных t2g-орбиталей, величина 10Dq(Δо) уменьшается (рис. 11.16, а). В случае лигандов сильного поля (например, СО, NO, CN–), t2g-орбитали которых расположены по энергии выше t2gорбиталей комплекса, величина 10Dq(Δо) увеличивается (рис. 11.16, б). В таких комплексах происходит перенос электронной плотности не только в прямом направлении (от лиганда к центральному атому), но и в обратном — от центрального атома к лиганду. Это дополнительное взаимодействие называют дативной π-связью.
Рис. 11.15. Схема МО для катиона гексаамминкобальта(III) [Co(NH3)6]3
Рис. 11.16. Зависимость параметра 10Dq от энергии π-орбиталей лиганда: а — энергия орбиталей лиганда ниже, чем у t2g-орбиталей металла;
б — энергия орбиталей лиганда выше, чем у t2g-орбиталей металла
Основные эмпирические параметры, которыми оперирует теория ПЛ, — параметр расщепления Dq и параметры Рака=15, характеризующие меж электронное отталкивание. Все эти параметры закономерно изменяются при варьировании природы центрального иона или лигандов; их абсолютные и относительные числовые значения хорошо описывают химию классических комплексных соединений.
При рассмотрении комплексов с позиций теории ПЛ большое значение имеет ковалентный вклад в связь металл—лиганд. При переходе от теории КП к теории ПЛ важное значение имеет параметр Полинга α, который характеризует степень смешения орбиталей металла и лиганда. Если электрон поровну принадлежит орбиталям обоих атомов связи (чисто ковалентный случай, когда электронная пара располагается строго посередине между атомами на линии связи), α = ±
. Другой крайний случай — чисто ионный, при котором взаимодействуют «точечные» заряды (именно его описывает теория КП); при этом α = 1. На практике оба этих крайних варианта встречаются редко.
Cтепень ионности (ковалентности) можно оценить по так называемому «нефелоксетическому эффекту» — увеличению объема d-орбиталей комплексообразователя из-за их экранирования неподеленными электронными парами лигандов. При этом межэлектронное отталкивание в комплексах оказываются меньшим, чем в соответствующих изолированных ионах металла. Усиление «нефелоксетизма» приблизительно соответствует возрастанию ковалентности связи лиганд—металл; наименьший нефелоксетический эффект оказывает лиганд F–; значительный нефелоксетический эффект у Вr– и CN–. Нефелоксетический эффект можно качественно характеризовать следующим рядом лигандов:
F– < О2– < ОН– < Н2О < ur < NH3 < en < С О2 24− < bipy < СО32− < < CN– < NCS– < Cl– < CNS– < acac < Br– < CNO– < N3– < I0 < S2– < I–
Количественно его определяют из соотношения:
β= Bкомплекс / Bион
где β — величина нефелоксетического эффекта, Bкомплекс — параметр Рака[ для комплекса, а Bион— для несвязанного иона.Параметр Рака[ Вкомплекс зависит от природы лигандов, основной фактор — ионный радиус лиганда. Чем больше ионный радиус, тем больше степень радиального смещения d-электронной плотности и тем меньше межэлектронное отталкивание (а значит, больше величина параметра В).
Теория ПЛ позволяет объяснить закономерности расположения лигандов в спектрохимическом ряду: лиганды располагаются в порядке уменьшения их σ- и π-осно[вности, т. е.в конце ряда находятся лиганды, которые проявляют в основном π-акцепторные свойства.
Следует отметить, что теорию ПЛ (наряду с теорией КП) можно использовать для разных целей:
-
предсказывать окраску и энергию электронных переходов (т. е. вид электронных спектров поглощения) неизвестных соединений, сравнивать устойчивость комплексов в растворах -
при интерпретации соответствующих электронных спектров поглощения предсказывать тип окружения (и КЧ) иона металла -
определять возможное число неспаренных электронов и магнитные свойства комплексов -
предсказывать некоторые структурные свойства кристаллических веществ, например взаимное расположение катионов в шпинелях -
детально изучать природу связи металл—лиганд.
Вопросы
11.17. Частицы [FеF6]4– и [Fе(CN)6]4– — высокоспиновый и низкоспиновый комплексы соответственно. Составьте энергетическую диаграмму образования связей в этих комплексах и опишите их магнитные свойства.
11.18. Параметр расщепления в поле лигандов 10Dq, определенный из спектроскопических данных, для некоторых комплексов переходных металлов имеет следующие значения:
| Комплекс | 10Dq (Δ), см–16 | Комплекс | 10Dq (Δ), см–1 |
| [СоВr4]2– | 2900 | [МnСl6]4– | 7500 |
| [СоСl4]2– | 3700 | [MnF6]4– | 8400 |
| [СоI4]2– | 2800 | [Mn(Н2O)6]2 | 8500 |
| [Мn(NCS)6]4– | 8800 | [NiВr6]4– | 7000 |
| [NiCl6]4– | 7200 | [NiF6]4– | 7300 |
| [Ni(Н2O)6]2 | 8500 | [Ni(NH3)6]2 | 10800 |
а) расположите лиганды в порядке возрастания способности к расще-
плению d-подуровней;
б) учитывая, что для комплексов Мn(II) и Со(II) энергия спаривания электронов составляет ∼20 000 см–1, определите, какие из перечисленных комплексов относятся к высокоспиновым.
11.19. Укажите возможную причину того, что магнитный момент иона [Мn(CN)6]3– почти в 2 раза меньше магнитного момента иона [МnF6]3–.
1 КИСТЯКОВСКИЙ Владимир Александрович (1865–1952) — российский и советский физикохимик. Работы по теории растворов, химической термодинамике и электрохимии.
2 ВЕРНЕР Альфред (1866–1919) — швейцарский химик. В 1891–1893 гг. предложил координационную теорию и в последующие годы осуществил ряд экспериментов, доказывающих ее правильность; разработал методы синтеза комплексов и определения их состава и строения. Лауреат Нобелевской премии по химии (1913).