Файл: Высокоэффективная жидкостная хроматография в фармацевтическом анализе.doc
Добавлен: 10.11.2023
Просмотров: 256
Скачиваний: 13
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Выбор неподвижной фазы имеет большое значение при проведении
любого хроматографического разделения. Синтез сорбентов для ионной
хроматографии затруднен, поскольку к ним предъявляется довольно много требований:
– сорбент должен иметь очень низкую ионообменную емкость (0,001-0,1 мэкв/г). Это связано с использованием кондуктометрического детектирования, при котором необходимы элюенты (растворы кислот, солей, оснований) с концентрацией менее 0,01 М. Для эффективного разделения такими разбавленными элюентами требуются низкоемкостные
ионообменные сорбенты;
– диаметр сорбента не должен превышать 50 мкм (обычно он равен 5–10 мкм). Только в этом случае можно достичь высокой эффективности разделения;
– зерна сорбента должны обладать высокой механической прочностью и устойчивостью к давлению, которое возникает приработе с мелкодисперсной неподвижной фазой;
– сорбент должен обладать высокой химической устойчивостью по отношению к элюирующему раствору. Он должен сохранять стабильность в широком интервале рН; Этим требованиям удовлетворяют поверхностно-пористые(пелликулярные) ионообменники, которые состоят из твердого инертного ядра, покрытого тонким слоем ионита. На таких сорбентах быстро устанавливается равновесие, поскольку диффузия в тонкую ионообменную пленку занимает мало времени. В результате ускоряется хроматографический процесс и достигается высокая эффективность разделения. Разделение катионов происходит на катионообменниках, которые содержат фиксированные группы SO3-, PO3,COO- и катионы в качестве противоиона. Равновесие ионного обмена описывается схемой:
Ионная хроматография весьма эффективный метод определенияионов, на рис. 1 и 2 показаны примеры разделения сложных смесей катионов и анионов. Ионная хроматография с кондуктометрическим детектором лучший метод определения неорганических анионов. Разделение проводят на ионообменниках низкой емкости (менее 0,1мМ/г) чаще всего поверхностно–модифицированных. Нижняя граница определяемых концентраций составляет 1-10 нг/л. Воспроизводимость по высотам и площадям : Sr не более 0,05. Наиболее часто ионную хроматографию используют для определения:[1]
– анионов неорганических кислот (HCl, HNO3, H2S, H3BO3 и др.);
– моно- и дикарбоновых кислоты;
– щелочных и щелочноземельных металлов;
– анионных комплексов переходных металлов;
– оксоанионов;
– алифатических аминов;
– оксидов азота, серы и фосфора.
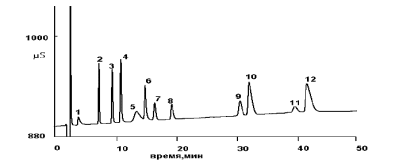
Рис. 1 Разделение смеси катионов на ионообменнике IonPac SCG 1: 1 –
медь; 2 – литий; 3 – натрий; 4 – аммоний; 5 – никель; 6 – калий; 7 – цинк; 8
– кобальт; 9 – марганец; 10 –магний; 11 – кальций; 12 – кадмий.
Подвижная фаза: смесь 4мМ винной и 2мМ щавелевой кислот
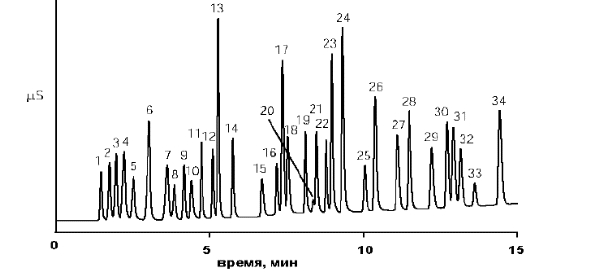
Рис.2 Разделение анионов на ионообменнике IonPac AG11: 1 – изопро-
пилэтилфосфонат, 2 – Quinate; 3 – фторид; 4 – ацетат, 5 – пропионат; 6-
формиат; 7 – метилсульфонат; 8 – Pyruvate; 9 – хлорат; 10 – валериановая
кислота; 11 – монохлорацетат; 12 – бромат; 13 – хлорид; 14 – нитрит; 15 –
трифторацетат; 16 – бромид; 17 – нитрат; 18 – перхлорат; 19 – селенит; 20
– карбонат; 21 – малонат; 22 – малеат; 23 – сульфат; 24 – оксалат; 25 – ке-
томалонат; 26 – SnO42-; 27 – фталат; 28 – фосфат; 29 – хромат; 30 – цитрат;
31 – трикарбалилат; 32 – изоцитрат; 33 – цис-ацинитат;34 – транс-
ацинитат; Подвижная фаза (50 –100) мМ NaOH, градиентный режим
ГЛАВА III. Оценка пригодности ВЭЖХ методик в фармацевтическом анализе
Валидация (оценка пригодности) методики анализа — это экспериментально обоснованное доказательство ее пригодности для получения результатов, имеющих достаточную точность и прецизионность. Валидации должны подвергаться методики анализа, применяемые при разработке и определении показателей и норм качества фармацевтической продукции. В случае использования методик, изложенных в государственных фармакопейных статьях, не требуется оценивать их пригодность, если анализ проводят в строгом соответствии с текстом фармакопейной статьи. В большинстве остальных случаев, особенно при изменении в составе готового лекарственного средства, изменении в синтезе лекарственной субстанции или изменении в самой аналитической методике, требуется оценка пригодности методики анализа.[2]
3.1. Тестирование аналитических методик, как единой аналитической системы
Единая аналитическая система предполагает, что оборудование, электроника, аналитические операции и анализируемые образцы составляют единую систему, которую можно исследовать как целое. В общем случае тестирование аналитической методики как единой аналитической системы дает возможность определять следующие валидационные характеристики: специфичность (Specificity), прецизионность (Precision), линейность (Linearity), точность (Accuracy), диапазон применения (область действия, Range), предел обнаружения (Detection Limit), предел количественного определения (Quantitation Limit), стабильность растворов (Solutions Stability)3. В зависимости от типа методики могут требоваться не все, а лишь некоторые из валидационных характеристик.
Специфичность методики-Для подтверждения специфичности вышеупомянутых методик обычно требуется показать, что пики определяемых веществ хорошо разделены между собой и хорошо отделены от пиков основных примесей, системных пиков (из растворителя образца) и пиков плацебо. С этой целью специфичность подтверждается набором хроматограмм, как минимум: а)испытуемого раствора, б)раствора субстанции определяемого вещества, в) растворителя образца (blank), г)плацебо — для лекарственных форм, д)раствора для определения пригодности хроматографической системы. В качестве критерия полноты разделения пиков обычно используют коэффициент разделения пиков Rs. Специфичность рекомендуется также подтвердить результатами исследования "чистоты " пика определяемого вещества. Для этой цели наиболее часто используют диодно-матричный детектор, с помощью которого по специальной программе проверяется спектральная однородность пика определяемого вещества. Принцип оценки "чистоты пика" заключается в следующем. Хроматограмму снимают одновременно при многих длинах волн (их количество — п). В каждой точке пика получают спектр, который математически характеризуется вектором, координаты которого в и-мерном пространстве представляют собой величины абсорбции (AU) при заданных (и) длинах волн (длина вектора пропорциональна концентрации вещества в анализируемом растворе). Различие между спектрами оценивают по величине угла между векторами спектров, называемого углом спектрального контраста G (Spectral Contrast angle). Если 9 равен нулю, то это означает, что спектры подобны (однородны), то есть величина абсорбции одного из спектров при любой длине волны
X может быть получена путем умножения на постоянный множитель величины абсорбции другого спектра при той же X. Для оценки спектральной однородности хроматографического пика в каждой из точек пика определяют угол спектрального контраста между вектором спектра в этой точке и вектором спектра в вершине пика 9, и находят максимальное значение Qp
Специфичность методик определения содержания примесей-Для подтверждения специфичности методик определения примесей в субстанциях требуется показать, что 1) методика дает возможность обнаруживать пики основных продуктов деструкции лекарственного вещества, 2) пики примесей достаточно хорошо разделены между собой и отделены от пика лекарственного вещества и системных пиков (из растворителя образца).
Для подтверждения специфичности методик определения примесей в лекарственных формах требуется показать, что 1) методика дает возможность обнаруживать пики основных примесей, содержание которых в лекарственной субстанции нормируется, 2) пики основных примесей достаточно хорошо разделены между собой и отделены от пика лекарственного вещества и системных пиков (из растворителя образца).
Если же основные примеси неизвестны или отсутствуют тоВ этом случае целесообразно использовать специальные эксперименты по химической модификации структуры лекарственного вещества, в ходе которых раствор лекарственного вещества (и/или само вещество) подвергают воздействиям, способствующим его частичной деструкции. При этом образуются примеси родственных соединений. Степень разложения вещества можно легко оценить по уменьшению площади основного пика на хроматограмме раствора, подвергнутого воздействию по сравнению с аналогичным пиком на хроматограмме исходного раствора. По разделению пиков примесей между собой и с пиком лекарственного вещества и по чистоте пика лекарственного вещества делают вывод о специфичности тестируемой методики.[2]
3.2.Диапозон определения методики
Диапазон применения методики — это интервал между наименьшей и наибольшей концентрациями (количествами) определяемого вещества, включая эти концентрации (количества), в котором:
1) соблюдается линейность
2) воспроизводимость не выходит за пределы допустимых (заданных) значений
3) методика является достаточно точной.
-
на практике достаточно доказать, что диапазон применения методики охватывает "с запасом" нормируемые границы содержания определяемых веществ, указанные в фармакопейных статьях. Для количественного определения основных веществ в субстанциях и лекарственных формах: от 80 до 120% от номинального содержания (т.е. концентрация испытуемых растворов должна охватывать диапазон не менее 80- 120% относительно концентрации раствора стандартного образца, используемого в тестируемой методике).
-
Для однородности дозирования: от 70 до 130 % от номинального содержания, если для испытания не требуется более широкий интервал (например, для аэрозолей).
-
Для испытаний на растворение: ± 20 % (абсолютных) от нормируемой величины высвобождения. Например, если при контроле высвобождения пролонгированных лекарственных средств нормируемая величина высвобождения составляет от 20 % за первый час и до 90 % за 24 ч, то диапазон применения должен быть от 0 до ПО % от номинального содержания.[2]
3.3. Предел обнаружения
Предел обнаружения (LOD) определяют как наименьшую концентрацию анализируемого вещества в образце, которую можно детектировать (обнаружить отклик) при заданных условиях теста . Для ВЭЖХ методик анализа субстанций и лекарственных форм предел обнаружения требуется находить для предельных тестов определения примесей. Очевидно, что предел обнаружения может зависеть от детекторов и насосов хроматографов, поэтому рекомендуется его оценить на различных приборах, включая те, которые могут быть у потребителя методики. Независимо от метода оценки предела обнаружения необходимо хроматограммой визуально подтвердить, что при концентрации, соответствующей пределу обнаружения определяемого вещества, действительно имеется его пик, явно превышающий шум базовой линии. В соответствии с Американской фармакопеей можно утверждать, что для методик, используемых в фармации, в подавляющем большинстве случаев нет необходимости определять фактический предел обнаружения определяемых веществ (исключение — методики контроля полноты очистки технологического оборудования). Достаточно показать, что примесь надежно детектируется на нормируемом уровне.
3.4. Предел количественного определения
Предел количественного определения (LOQ) — наименьшая концентрация аналита, которая может быть определена с приемлемой прецизионностью (воспроизводимостью) и точностью при регламентированных условиях метода.ICH и USP рекомендует несколько способов оценки LOQ для методик определения содержания примесей:[2]
Оценка предела количественного определения LOQ по соотношению "Сигнал/Шум" S/N' = 10.
Это наиболее распространенный способ. Однако, поскольку в нем используются высоты откликов (а не их площади), он более подходит для методик ВЭЖХ, в которых в качестве отклика применяются высоты пиков. Однако при этом не учитываются требования к воспроизводимости методики ВЭЖХ.