ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 30.10.2019
Просмотров: 1807
Скачиваний: 2

6
Journal of Pharmacological Sciences
©2005 The Japanese Pharmacological Society
Survey Review
J Pharmacol Sci
99
, 6 – 38 (2005)
The Kallikrein-Kinin System: Current and Future Pharmacological
Targets
Marie Eve Moreau
1
, Nancy Garbacki
2
, Giuseppe Molinaro
1
, Nancy J. Brown
3
, François Marceau
4
,
and Albert Adam
1,
*
1
Faculty of Pharmacy, University of Montreal, 2900 boul. Édouard-Montpetit, C.P. 6128, Succursale Centre-Ville,
Montréal (Québec), Canada H3C 3J7
2
Laboratory of Human Physiology, University of Liège, 3 Avenue de l’Hôpital, Sart Tilman, Belgium B-4000
3
Division of Clinical Pharmacology, Department of Medicine, School of Medicine, Vanderbilt University,
60 Robinson Research Building (RRB), Nashville, TN 37232-6602, USA
4
Centre de Recherche en Rhumatologie et Immunologie, CHUL-CHUQ, 2705 boul. Laurier,
Québec (Québec), Canada, G1V 4G2
Received May 24, 2005
Abstract.
The kallikrein-kinin system is an endogenous metabolic cascade, triggering of which
results in the release of vasoactive kinins (bradykinin-related peptides). This complex system
includes the precursors of kinins known as kininogens and mainly tissue and plasma kallikreins.
The pharmacologically active kinins, which are often considered as either proinflammatory or
cardioprotective, are implicated in many physiological and pathological processes. The interest
of the various components of this multi-protein system is explained in part by the multiplicity of
its pharmacological activities, mediated not only by kinins and their receptors, but also by their
precursors and their activators and the metallopeptidases and the antiproteases that limit their
activities. The regulation of this system by serpins and the wide distribution of the different
constituents add to the complexity of this system, as well as its multiple relationships with other
important metabolic pathways such as the renin-angiotensin, coagulation, or complement path-
ways. The purpose of this review is to summarize the main properties of this kallikrein-kinin
system and to address the multiple pharmacological interventions that modulate the functions of
this system, restraining its proinflammatory effects or potentiating its cardiovascular properties.
Keywords
: kallikrein-kinin system, B
1
and B
2
receptors, metallopeptidase, pharmacological agent
Introduction
............................................................. 7
Part I: The Kallikrein-Kinin System
..................... 8
I- Kinins
.............................................................. 8
1. The kininogens: precursors of kinins
......... 8
1.1 High-molecular-weight kininogen
1.2 Low-molecular-weight kininogen
2. The kinin-forming systems
.......................... 9
2.1 The plasma kinin forming system
2.1.1 Plasma prekallikrein
2.1.2 Contact system activation of plasma
2.2 Endothelial cells and kinin forming activity
2.3 Tissue kallikrein-kinin system
2.4 Other kinin forming enzymes
3. Regulation of the kininogenase activity
...... 10
II- Metabolism of Kinins
................................... 11
1. Angiotensin I-converting enzyme (ACE)
.... 11
1.1 Definition
1.2 Synthesis, regulation, and localization
1.3 Properties
1.3.1 Angiotensinase vs kininase
1.3.2 ACE GPIase activity
1.3.3 ACE: a signal transduction molecule
1.3.4 ACE insertion / deletion polymorphism
*Corresponding author.
FAX: +1-514-343-2102
E-mail: albert.adam@umontreal.ca
Invited article

The Kallikrein-Kinin System
7
2. Neprilysin
.....................................................
12
2.1 Definition
2.2 Synthesis, localization, and properties
3. Aminopeptidase P
.........................................
13
3.1 Definition
3.2 Synthesis, regulation, and localization
3.3 Properties
4. Carboxypeptidase N and M
..........................
13
5. Other peptidases
..........................................
13
III- Kinin Receptors
........................................... 14
1. Pharmacological classification
....................
14
1.1 Potency order of agonists
1.2 Affinity of antagonists
2. Molecular classification
...............................
17
2.1 Organization and structure
of the receptor genes
2.2 Receptors expression and regulatory
elements in gene promoters
2.3 Second messengers
2.4 Receptor desensitization
Part II: The Kallikrein-Kinin System:
Pathophysiology and Pharmacological
Target
......................................................... 19
I- The Kinin Forming System in Plasma
......... 19
1. Genetic defects
..............................................
19
1.1 Defects of the contact system components
1.2 Defect in the control of the contact system:
C1 inhibitor
1.2.1 Definition
1.2.2 Pathophysiology
1 2.3 Treatment of hereditary angioedema
1.2.3.1 Serine proteases inhibitors
1.2.3.2 DX88
1.2.3.3 Attenuated androgens: Danazol
®
,
Stanozolol
®
1.2.3.4 Antifibrinolytic drugs: tranexamic
acid (Transamin
®
, Cyklokapron
®
,
Exacyl
®
, Cyklo-f
®
)
1.2.3.5 B
2
-receptor antagonists: HOE 140
(Icatibant
®
or JE049
®
)
2. Acquired diseases
.........................................
21
2.1 Sepsis
2.2 Anaphylactoid and severe hypotensive
reactions
3. Antithrombolytic treatment and kinins
.......
21
II- The Metabolism of Kinins
............................ 21
1. ACE: pathophysiology
.................................
21
2. ACE inhibitors (ACEi)
.................................
21
2.1 Multicenter clinical randomized trials
2.2 Role of bradykinin in the cardiovascular
effects of ACEi
3. Vasopeptidase inhibitors (VPi)
....................
22
3.1 Multicenter clinical randomized trials
3.2 Role of kinins in the cardiovascular effects
of VPi
4. Kinins and side effects of metallopeptidase
inhibitors
....................................................... 23
III- Kinin Receptors
........................................... 24
1. Receptor polymorphisms and pathology
.....
24
1.1 Polymorphism of B
2
receptors
1.1.1 +9 /
−
9, exon 1 polymorphism
1.1.2 (C
−
58
→
T), promoter region
polymorphism
1.1.3 (C
181
→
T), exon 2 polymorphism
1.2 Polymorphisms of B
1
receptors: (G
−
699
→
C)
substitution
2. Kinin receptors as pharmacological
targets
...........................................................
25
2.1 Cardiovascular and renal diseases
2.2 Inflammation
2.3 Pain and neurological applications
2.4 Diabetes
2.5 Renal disease
2.6 Airway disease
2.7 Angioedema
Conclusion
............................................................... 28
Introduction
The existence of the kallikrein-kinin system was first
discovered almost one century ago when Abelous and
Bardier, in 1909, showed the hypotensive effect of
human urine (1). Since that time, this system has been
and continues to be the subject of intensive research.
In fact, more than 30,000 papers are referenced for
kallikrein-kinin in Medline for the last 50 years.
Such a scientific interest is explained in part because
of the duality and the complexity of this system. Brady-
kinin (BK) is at the center of this system; however, it is
not the only pharmacologically active kinin. The kinins
(BK-related peptides) are generated from two types of
kininogens, mainly by two types of activators: tissue and
plasma kallikreins. In fact, two classes of kinin receptor
ligands are now recognized corresponding to each
receptor subtype (B
1
and B
2
receptors). The expression
of these receptors is regulated by specific mechanisms.
The duality also exists as to the pharmacological activity
of kinins, which are often considered as either pro-
inflammatory or protective (namely for heart, kidney
function, angiogenesis-promoting) depending on the
experimental approach, or scientific interest. This
system is also complex in its distribution, as the different
constituents have been shown to be present in plasma,

ME Moreau et al
8
but also on blood cells, in various tissues or their
exocrine secretions. The autocrine or paracrine activity
of kinins is regulated by several metallopeptidases, the
relative importance of which varies from one biological
medium to the other. Finally, the regulation of this
system by serpins adds to the complexity of the system,
as well as its multiple relationships with other important
metabolic pathways such as the renin-angiotensin,
coagulation or complement pathways.
An understanding of the multifaceted aspects of the
different constituents of this system is necessary to grasp
the complexity of its multiple pharmacological acti-
vities, mediated not only by kinins and their receptors,
but also by their precursors and their activators, and the
metallopeptidases and the antiproteases that limit their
activities.
The purpose of this review is, first, to summarize
the main properties of the various constituents of this
complex but fascinating system. We will secondly
address the multiple pharmacological interventions that
modulate the functions of the kallikrein-kinin system
and their clinical applications.
Part I: The Kallikrein-Kinin System
The kallikrein-kinin system represents a metabolic
cascade that, when activated, triggers the release of
vasoactive kinins. This complex multi-protein system
includes the serine proteases tissue and plasma
kallikreins, which liberate kinins from high- and low-
molecular-weight kininogen (HK and LK). Kinins exert
their pharmacological activities by binding specific
receptors, before being metabolized by various pepti-
dases.
I- Kinins
In humans and in most mammals, the term “kinin”
refers to the nonapeptide, BK (Arg-Pro-Pro-Gly-Phe-
Ser-Pro-Phe-Arg), the decapeptide kallidin (KD: Lys-
BK), and their carboxy-terminal des-Arg metabolites.
Other kinins, like T-kinin (Ile-Ser-BK) and Met-T-kinin
have only been reported in the rat (2).
Kinins are implicated in many physiological and
pathological processes. By virtue of their ability to
activate endothelial cells, leading to vasodilation, in-
creased vascular permeability, tissue-type plasminogen
activator (t-PA) release, production of nitric oxide (NO),
and mobilization of arachidonic acid, they participate in
physiological (regulation of blood pressure, renal and
cardiac functions) and pathological processes like
inflammation.
1. The kininogens: precursors of kinins
HK and LK, the precursors of kinins (BK and KD),
are produced from a structural gene localized to chromo-
some 3q26
→
qter that is thought to have originated as a
result of two successive duplications of a primordial
kininogen gene (3, 4). This gene consists of 11 exons.
The first 9 exons encode the heavy chain. The 10th exon
codes for BK and the light chain of HK; the light chain
of LK is coded by exon 11.
Both HK and LK have an identical aminoacid
sequence starting at the N-terminus (heavy chain) and
continuing to 12 aminoacids beyond the BK moiety but
differ at the C-terminal because of alternative splicing,
thereby providing the two kininogens with different
light-chain moieties (3, 4). In fact, both native proteins
are produced as single chain polypeptides, and the light
or heavy chain nomenclature refers to their disulfide
bond-assembled structure after activation by kallikrein
cleavage.
1.1 High-molecular-weight kininogen (HK)
HK, an
α
-globulin, circulates in plasma as an 88- to
120-kDa single-chain glycoprotein at a concentration of
70 to 90
µ
g
/
mL (5). This kininogen is a multifunctional
protein composed of six domains. Each domain is
thought to have distinct functions. HK heavy chain
(64 kDa) contains domains 1, 2, and 3. The light chain
(45 to 58 kDa) is comprised of domains 5 and 6. The
heavy chain and light chain are linked by domain 4,
which contains the BK sequence. Domain 1 has a low
affinity calcium-binding site (6), domains 2 and 3 have
specific sequences (Gln-Val-Val-Ala-Gly) that inhibit
cysteine proteases (7), and domain 3 has platelet- and
endothelial cell-binding activity (8). Domain 5 has cell-
binding sites, antiangiogenic properties, and sequences
for heparin binding (9 – 11). HK binds to negatively
charged surfaces through the histidine region of the light
chain corresponding to domain 5. Domain 6 has
prekallikrein and factor XI-binding sites (12). The
ability to bind to a surface (domain 5) and simulta-
neously complex factor XI or prekallikrein (domain 6) is
responsible for the cofactor activity of HK in contact
activation of plasma (13).
1.2 Low-molecular-weight kininogen (LK)
LK is a
β
-globulin present in human plasma at a
concentration between 170 and 220
µ
g
/
mL (5). It has a
molecular mass ranging from 50 to 68 kDa (5, 14). LK
has the same basic structure as HK: the same amino-
terminal heavy chain of 50 to 60 kDa linked to a
carboxy-terminal light chain by the kinin segment (14).
The light chain of LK is only 4 to 5 kDa and lacks
contact activation and prekallikrein-binding sites. The
The Kallikrein-Kinin System
9
function(s) of the light C-terminal chain of LK remain
unknown.
2. The kinin-forming systems
Classically, there are two main pathways by which
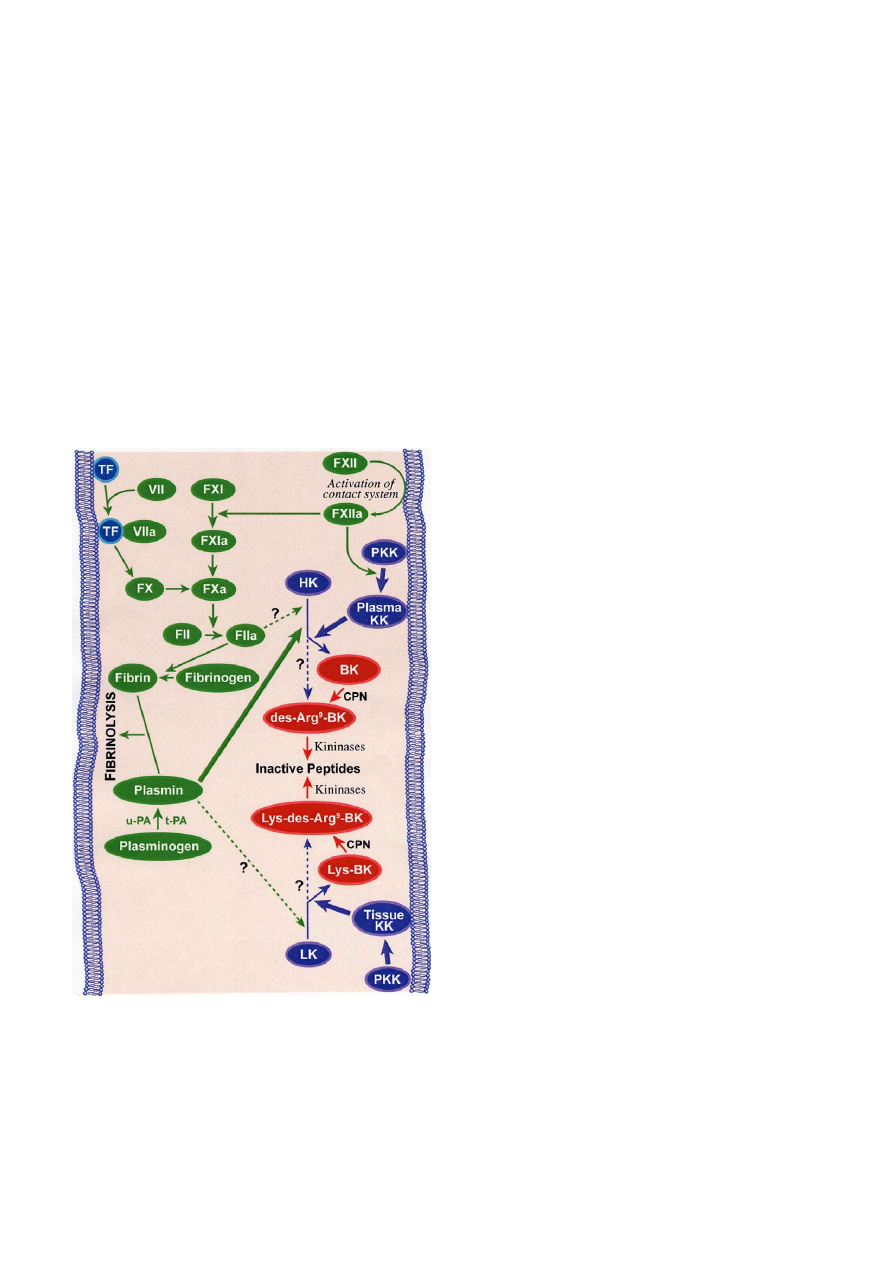
kinins are generated (Fig. 1). The plasma kallikrein-
kinin system, by far the more complex, initiates acti-
vation of the intrinsic coagulation pathway. The second
and simpler pathway of kinin generation involves tissue
kallikrein and its substrate, LK. Each of these enzyme
systems may play different pathophysiological func-
tional roles.
2.1 The plasma kinin forming system
The plasma kinin forming system, also called the
contact system of plasma, consists of 3 serine pro-
enzymes (factor XII or Hageman factor, factor XI, and
prekallikrein) and the kinin precursor HK.
2.1.1 Plasma prekallikrein
Chromosomal localization of the human plasma
kallikrein gene was mapped to the q34 – q35 region of
the long arm of chromosome 4 (15). Plasma kallikrein
(EC 3.4.21.34), a serine protease, is encoded by a single
gene,
KLKB1
, and synthesised in the liver. It is pre-
dominantly secreted by hepatocytes as an inactive
molecule called prekallikrein that circulates in plasma as
a heterodimer complex bound to HK with 1:1 molar
stoichiometry (16, 17). Prekallikrein is a single chain
α
-globulin that is present in the plasma of humans and
of other animal species at a concentration of 35 –
50
µ
g
/
mL. About 80 – 90% of prekallikrein is normally
complexed to HK (16, 18).
2.1.2 Contact system activation of plasma
Contact of plasma with a negatively charged surface
leads to the binding and autoactivation of factor XII
(Hageman factor) to factor XIIa, activation of pre-
kallikrein to kallikrein by factor XIIa, and cleavage of
HK by kallikrein to release BK (19). Factor XII acti-
vation is not only a first step in the initiation of the
intrinsic clotting cascade and the generation of kinins,
but it also leads to the activation of the complement
pathway (20).
In vitro, non-physiologic substances, such as glass
(negatively charged silicates), carrageenan, kaolin, and a
sulfated polysaccharide dextran sulfate (21) activate
the contact system of plasma. In vivo, the physiologic
surface remains unknown. Pathologic initiators may
include proteoglycans (sulfate residues on heparin
sulfate or chondroitin sulfate or mast-cell heparin).
Endotoxins (lipopolysaccharide (LPS)) and crystals of
uric acid or pyrophosphate (22) have also been hypo-
thesized to be pathological activators. This intrinsic
coagulation
/
kinin-forming cascade appears to be in
equilibrium in plasma even in the absence of any exo-
genous surface. That is, activation occurs continuously
at a finite rate, but is held in check by plasma inhibitors
(19).
Plasma kallikrein cleaves human HK in a two-step
process. First, HK is cleaved at the Arg
389
-Ser
390
bond of
the carboxy-terminal portion of the BK sequence,
leaving the BK attached to the carboxy-terminal end of
the heavy chain, and the sequence Leu
378
-Met-Lys-
Arg
381
is cleaved at the Lys
380
-Arg
381
bond to liberate
BK from the heavy chain (23). As a result of domain
rearrangement, HKa acquires new properties. Recent
observations suggest that HKa inhibits endothelial cell
Fig. 1.
The kinin-forming systems. The kallikrein-kinin system and
its interactions with both intrinsic and extrinsic coagulation cascades
and fibrinolysis. Solid lines are established pathways, whereas
dashed lines are speculative or experimental activation pathways.
TF: tissue factor; PKK: prekallikrein; HK: high-molecular-weight
kininogen; LK: low-molecular-weight kininogen; BK: bradykinin;
CPN: carboxypeptidase; t-PA: tissue plasminogen activator; u-PA:
urokinase plasminogen activator.

ME Moreau et al
10
proliferation and neovascularisation due to antiapoptotic
properties. This property contrasts with the angiogenic
effect of HK and LK, due to the release of BK (24).
2.2 Endothelial cells and kinin forming activity
Another mechanism for initiation of the activation of
the kallikrein-kinin system depends on binding of com-
ponents of the contact activation cascade on the surface
of cells as leukocytes, platelets, endothelial cells, and
myocytes (25).
HK specifically binds to platelets, granulocytes, and
endothelial cells in a zinc-dependent, saturable and
reversible reaction (26, 27). The dissociation constant
equals 15 nM, indicating high affinity binding (21). This
binding involves both the heavy (domain 3) and light
(domain 5) chains of HK (28), which could be
considered as a receptor for prekallikrein on endothelial
cells (29 – 31). Binding of HK to endothelial cells leads
to activation of prekallikrein to kallikrein (11, 25, 31)
and presumably a release of BK from HK (31, 32).
The interaction of HK with endothelial cell mem-
branes involves a multiprotein receptor complex com-
prising at least cytokeratin 1, gC1qR, and the urokinase
plasminogen activator receptor (u-PAR) (33 – 38).
These three proteins co-localize on the endothelial cell
membrane (39). The same three proteins form a receptor
complex for factor XII, but binding of factor XII in
vivo is likely limited both by the low plasma concentra-
tion of free Zn
2+
, which is below the requirement for
factor XII binding, and by the much higher plasma
concentration of HK (40).
2.3 Tissue kallikrein-kinin system
Tissue (glandular) kallikrein (EC 3.4.21.35) is an
acid glycoprotein which differs from plasma kallikrein.
This tissue serine protease is encoded by one of the
kallikrein gene family,
KLK1
gene, located on
chromosome 19 locus q13.2 – q13.4 (41, 42). Tissue
kallikrein is widely distributed (kidney, blood vessels,
central nervous system (CNS), pancreas, gut, salivary
and sweat glands, spleen, adrenal, and neutrophils) (17,
42), and this wide distribution suggests a paracrine
function (43). The origin of tissue kallikrein detected
in plasma has been suggested to be the exocrine glands.
Tissue kallikrein is synthesized as a proenzyme, pro-
kallikrein, which is inefficiently activated by plasmin or
plasma kallikrein (44).
Tissue kallikrein releases KD from LK (42), cleaving
the Met
379
-Lys
380
and Arg
389
-Ser
390
bonds (45). Although
LK is considered to be the main substrate of tissue
kallikrein, tissue kallikrein is also capable of cleaving
HK.
2.4 Other kinin forming enzymes
In addition to tissue and plasma kallikrein, other se-
rum and tissue proteases have a kinin forming capacity
(46). Plasmin, which is responsible for lysis of the fibrin
clot, releases not only BK but also des-Arg
9
-BK from
HK (47), circulates in plasma as an inactive zymogen,
plasminogen. The presence of plasminogen activators
and their inhibitors is essential in controlling fibrinolysis
(48).
The major activator of plasminogen in vivo is tissue
plasminogen activator (t-PA), a serine protease synthe-
sized and secreted by endothelial cells as a single chain
active enzyme (46). In the absence of fibrin, t-PA is an
inefficient activator of plasminogen but the binding of
t-PA to fibrin greatly accelerates the activation of
plasminogen. Urokinase or urinary type plasminogen
activator (u-PA) can also activate plasminogen and plays
an important role in the degradation of the extracellular
matrix. This facilitates the migration of cells which is
important in wound healing and in tumour invasion
and metastasis. u-PA can be of primary importance for
cell-mediated activation of plasminogen in tissues,
whereas t-PA, possessing a high affinity for fibrin, can
be of primary importance for the lysis of fibrin clots in
the circulation (49).
Factor XIIa, XIa, and kallikrein are also capable of
converting plasminogen to plasmin in vitro. The contri-
bution of these enzymes to the activation of plasminogen
in vivo is uncertain; in fact, deficiencies in these proteins
do not appear to lead to pathological states that could
be explained by an impaired fibrinolysis (48).
3. Regulation of the kininogenase activity
Protease inhibitors regulate the contact activation of
plasma. The serpins of plasma are namely C1-inhibitor
(C1INH), antithrombin III,
α
2
-macroglobulin,
α
1
-pro-
tease inhibitor, and
α
2
-antiplasmin (19, 50). However,
C1INH is the major regulator of the intrinsic system,
interfering with the activities of factor XIIa and of
kallikrein (51). Both C1INH and
α
2
-macroglobulin
account for more than 90% of the kallikrein inhibitory
activity of plasma.
The regulatory mechanism of tissue kallikrein remains
partly unknown. Kallistatin and aprotinin are an example
of serine protease inhibitors. Unlike many other serpins
that are present only in plasma, kallistatin can be
detected in various tissues, cells, and fluids, where it
regulates the activity of tissue kallikrein (52, 53).
Plasmin, t-PA, and u-PA are also regulated by inhi-
bitors in the serpin family. The primary inhibitor of
active plasmin is
α
2
-antiplasmin. Similarly, anti-
thrombin,
α
1
-antitrypsin, and C1INH have been shown
to inhibit plasmin in vitro but have a minimal physio-