ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 30.10.2019
Просмотров: 1820
Скачиваний: 2

ME Moreau et al
16
Table 2.
Pharmacological and clinical application of kinin B
2
-receptor ligands
Ligands
Appllication
Studies
References
Agonists
Labradimil ([Hyp
3
, Thi
5
, 4-Me-Tyr
8
Ψ
(CH
2
-NH)Arg
9
]-BK)
Vascular permeability (blood brain barrier):
adjuvant to chemotherapy of brain tumors
In vivo rodent models
Human: phase II studies on glioma
284
FR190997
Hypertension
Rat
283
Antagonists
First generation
[
D
-Phe
7
]-BK
Low potency,
Antagonist
/
partial agonist activity
Rat uterus, guinea pig ileum
Rat
133
[Thi
5,8
,
D
-Phe
7
]-BK
Potent antagonist, no agonist activity
Rat uterus, guinea pig ileum
133
Second generation
HOE 140 (Icatibant;
D
-Arg-[Hyp
3
, Thi
5
,
D
-Tic
7
, Oic
8
]-BK)
High affinity, long-lasting, competitive
activity in but measurable affinity for B1R
Animal models (high affinity for the human,
rabbit, and guinea pig B2R)
134
No residual agonist effects
Resistance to peptidases
Acute rhinitis
Human, nasal treatment
325
Asthma
Human
269
Early stage of inflammation
Rat
317
Persistent inflammatory pain
Third generation – Nonpeptide compounds
Phosphonium family:
WIN64338
Inactive
On human tissues
137
Limited affinity
For guinea pig B2R
136
WIN62318
Micromolar binding affinity to human B2R
Identification of the absolute requirement
for B2R binding affinity: presence of two
positive charges at a distance of about 10 Å
separated by a lipophilic residue, playing the
role of Phe
8
side chain in the native ligand
138
Quinoline and imidazol
[1,2-
α
]pyridine family:
High B2R affinity and selectivity versus B1R
Oral activity at doses ranging between 1
and 30 mg
/
kg in different tests and species
282, 326
FR165649, FR173657, FR184280
Oral activity on hyperalgesia and inflammation
Rat, mice
138
FR167344
Selective and high potent binding activity
Guinea pig ileum, human A-431 cells
327
Bronchoconstricition
Guinea pigs (oral activity)
Designed as clinical candidate to treat
inflammatory diseases
Compound 38
High affinity
Human B2R
328
CP2522
High affinity
Modeled on CP0597 by replacing
β
-turn
conformation of the peptide by a rigid
1,4-piperazine ring
Human B2R
138
Substituted 1,4-dihydropyridines
B2R antagonist at the nanomolar range
Human B2R
138
Bradyzide
Hypertension Inflammation
Rodent, orally active, less potent
in human B2R
292
Natural compounds
Pyrroloquinoline alkaloid: Martinelline
Affinity for both B1R and B2R at the
micromolar range but not selective
Alkaloid isolated from the South American
tropical plant
Martinella iquitosensis
144
L-755807
Inhibition of BK binding to cloned human
B2R at micromolar range
Complex metabolite isolated from a culture
of the mould
Microsphaeropsis
sp.
No further pharmacological data
329

The Kallikrein-Kinin System
17
2. Molecular classification
Distinct B1R and B2R genes coexist in the human
genome and, most probably, in the genome of most if
not all mammals (129). It is interesting to note that a
fish and a bird (the chicken) possess only one type of
receptor for kinin homologs and that it is most related
to the mammalian B2R, which appears to be ancestral
to the B1R (129). Furthermore, the Human Genome
project revealed that the human kinin receptor genes are
clustered in tandem in the same locus of chromosome 14
(14q32.1 – q32.2), with less than 20 kb of genomic DNA
separating them. This has profound implications for the
study of genetic polymorphisms, as a genetic marker in
one of the two genes may point out a functional
alteration of the other, as this genetic distance is very
small and as both genes are likely to be transmitted
together vertically (see below).
The molecular definition of the kinin receptor was
initiated by the expression cloning of the rat B2R (145)
followed by that of the human B1R (146). The degree of
aminoacid identity is not very high between the human
B1R and B2R sequences (36%), but they are the most
highly related pair, followed by the receptors for angio-
tensin. The determination of the pharmacological profile
of cloned receptors from various species has ended
much speculation about the existence of multiple addi-
tional receptor subtypes, as it was recognized that the
rather large differences in their affinity to agonists and
antagonists were a species-related issue.
2.1 Organization and structure of the receptor genes
The B1R protein (40.4 kDa) exhibits a seven trans-
membrane structure typical for GPCR (G
α
q
and G
α
i
) and
possesses three consensus sites for N-linked glycosyla-
tion in extracellular domains, DRY (Asp-Arg-Tyr) and
NPXXY (Asn-Pro-XX-Tyr) motifs, and putative sites
for phosphorylation and acylation. B1R is not expressed
in significant levels in normal tissues (130). The expres-
sion of B1R is inducible rather than constitutive.
However, exceptions occur in mammals concerning the
inducible behavior of B1R. For example, dogs and cats
constitutively express the receptor (130).
The human B1R gene (
BDKRB1
) is located on
chromosome 14q32.1 – q32.2. The gene product con-
sists of 353 aminoacids and approximately 70% of its
overall genomic sequence is homologous to the mouse
and the rat
BDKRB1
genes. The three-exon structure
of the human B1R gene has been determined with the
protein sequence being encoded by exon 3 exclusively
(147, 148). Critical receptor epitopes for G-protein
binding and activation are located on multiple intra-
cellular domains, which are thought to act in concert
to form a binding site (149).
As B1R, the B2R protein structure is typical of that
of a GPCR consisting of a single polypeptide chain
that spans the membrane seven times, with the amino
terminus being extracellular (N-terminal domain) and
the carboxyterminus (C-terminal domain) being intra-
cellular and with three extracellular loops (EL1 – 3) and
three intracellular loops (IL1 – 3). Three consensus
sites for N-linked glycosylation are found in extra-
cellular domains. Moreover, the protein contains motifs
such as DRY and NPXXY partially embedded in cyto-
solic receptor domains that are common to most
rhodopsin family GPCRs (150), and the C-terminal tail
contains serine and threonine residues that are putative
phosphorylation sites and cysteines that are putative
sites for acylation.
The B2R has been identified in most tissues and is
particularly present on endothelial cells, smooth mus-
cular cells, fibroblasts, mesengial cells, some neurons,
astrocytes, and polynuclear neutrophils (151). Its gene
expression level is constitutive. The human
BDKRB2
gene product consists of 391 aminoacids and the
three-exon structure of the gene is also located on
chromosome 14 (152) but about 12 kb upstream from
BDKRB1
(153). About 80% of the human
BDKRB2
gene is homologous to the mouse and rat
BDKRB2
gene.
The human
BDKRB1
and
BDKRB2
are also similar to
each other with 36% genomic sequence homology. The
mRNA coding for the B2R (4 kb) is large compared
with that of B1R (1.4 kb).
2.2 Receptors expression and regulatory elements in
gene promoters
Expression of the B1R is up-regulated following some
types of tissue injury, and its relationship to pathology
is therefore obvious. Exposure to bacterial endotoxins
or cytokines, tissue trauma, inflammation, anoxia (130),
and myocardial infarction (154) are examples of induc-
ing stimuli. IL-1
β
and tumor necrosis factor (TNF)-
α
induce the expression of the B1R in vitro (155, 156)
and in vivo (157, 158).
Cytokine-induced B1R expression is mediated by
specific MAP-kinase pathways (notably, p38 and JNK)
and nuclear factor-
κ
B (NF-
κ
B) (159 – 162). Inflam-
matory induction of B1R expression correlates with
NF-
κ
B stimulation in various systems (163, 164).
Studies on the human embryonic IMR-90 cell line led
to the hypothesis of autoregulation of the B1R; stimula-
tion of either the B1R or B2R in these cells led to the
increased expression of the B1R (mRNA, protein) (165,
166). However, this model may not be generally appli-
cable, as these permissive cells produce autocrine IL-1
β
in response to the stimulation of various GPCRs (167)
and as kinin receptor stimulation does not up-regulate

ME Moreau et al
18
B1R expression in primary vascular cells (cultured
smooth muscle cells) or in vivo, following the activation
of the contact system in rabbits using dextran sulfate
(168). Evidence of the role of the transcription factor
AP-1 in B1R expression has been derived from a
construction composed of the 1.8 kb core promoter,
exon 1, and 1.5 kb of intron 1, exon 2, intron 2, and a
luciferase reporter (169).
The expression of B2R is up-regulated not only by BK
but also by cAMP and phorbol esters (170). Multiple
potential binding sites have been identified recently in
the B2R promoter (171): GATA-1, CCAAT displace-
ment protein, E2F, Egr2, IL-6 activator protein, NF-
κ
B,
p53, estrogens. IL-1
β
increases both the number of B2R
as well as the level of B2R mRNA through a prostanoid
and cAMP dependent pathway, which may lead to pro-
tein kinase A activation of the transcription factor cAMP
response element-binding protein (172).
2.3 Second messengers
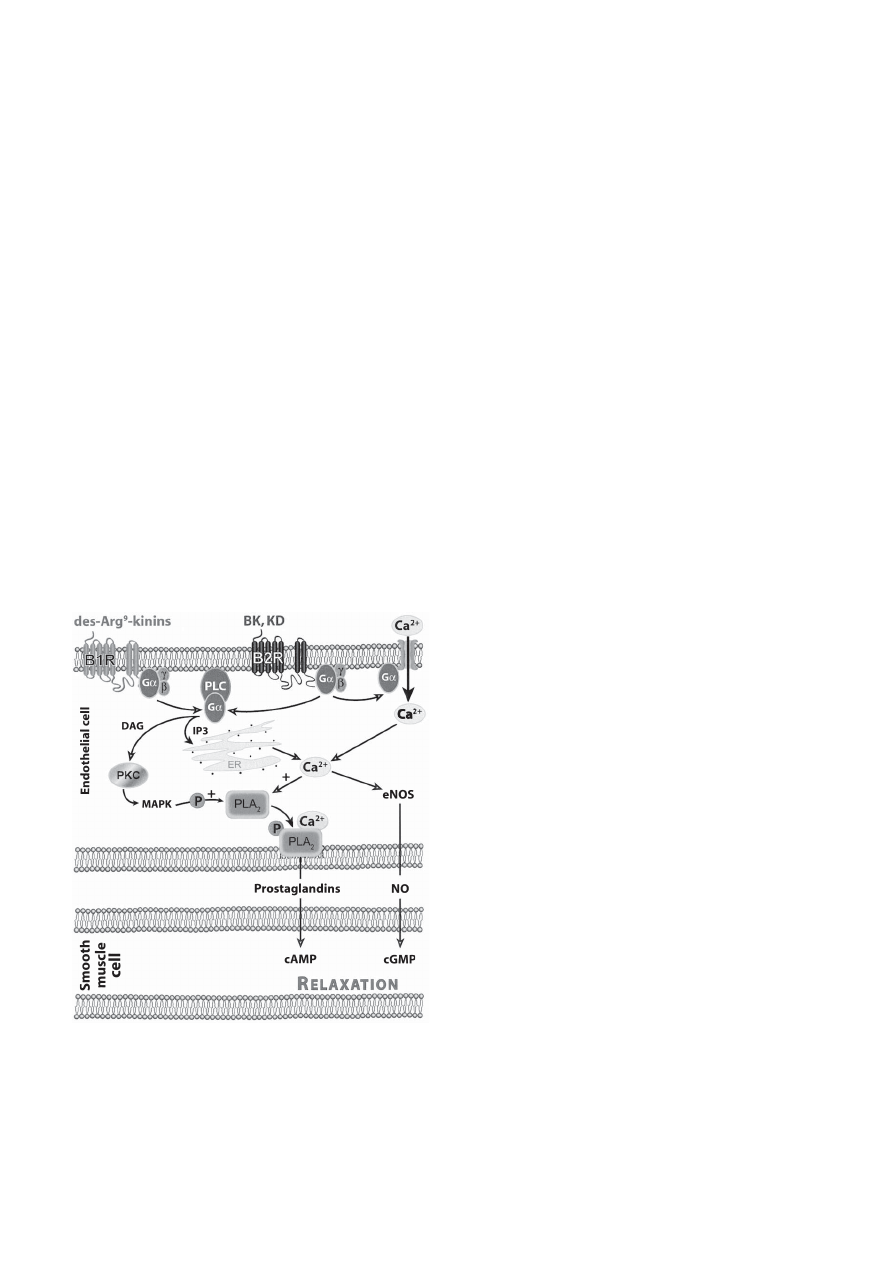
Signal transduction of kinin receptors activates
several second messenger systems, depending on
cellular type, via the activation of G-proteins (Fig. 2).
Activation of adenylyl cyclase or guanylyl cyclase is a
transduction mechanism that leads to the production of
cAMP and to cGMP, an efficient vasodilatory mecha-
nism in vascular smooth muscle. However, the kinin
receptors, coupled to G
q
and G
i
proteins, are only indi-
rectly linked to cyclic nucleotide signaling: via the
endothelial production of NO, itself capable of acti-
vating the soluble guanylate cyclase in neighboring cell
types or via the production of prostaglandins (PGs) I
2
or
E
2
that possess G
s
-protein-coupled receptors. The acti-
vation of ionic channels and of PLs A
2
, C, and B also
play a role in kinin receptor signaling. PLC products,
inositol 1,4,5-triphosphate and diacylglycerol (DAG)
(173), are respectively responsible for transporting
calcium into the cytosol from intracellular stores and
for protein kinase C translocation from the cytosol to
the plasma membrane. Ca
2+
mediates the activation of
endothelial NO synthase (eNOS) and ultimately the
production of NO in endothelial cells (174) and stimu-
lation of PLA
2
(129). Arachidonic acid could also be
liberated from cellular membrane phospholipids by the
action of PLA
2
and lead to PG production. In addition
to these classical pathways, B2R can activate signaling
proteins that possess cytoplasmic tyrosine kinase activity
(175). The activated B2R also transiently promotes
tyrosine phosphorylation of MAP-kinase (176), PLC
(177), and Hsp90 (178) and has a direct interaction
with neuronal and eNOS allowing the control of NOS
phosphorylation and NO production (179, 180). Further-
more, BK activates caveolae-associated Janus-activated
kinase
/
signal transducers and activators of the transcrip-
tion (JAK
/
STAT) pathway in endothelial cells (181).
Tyrosine kinases of the JAK family phosphorylate
STAT proteins that directly regulate transcription of
specific genes.
B1R also interacts directly with G
α
q
and G
α
i
proteins
and recruits essentially the same signaling pathways as
the B2R. Although the B1R and B2R seem to couple
to similar cellular signal transduction pathways, the
patterns of signaling are different in terms of variation
of Ca
2+
concentration (in duration and in intensity,
see below). B1R is principally associated with PLC
activation and with the phosphoinositol pathway, but
also acts through PLA
2
and the MAP-kinase (130).
2.4 Receptor desensitization
Besides their pharmacological differences, B1R and
B2R also display important differences in their suscepti-
bility to desensitization. When activated by an agonist,
B2R undergoes to a rapid desensitization (182), involv-
ing phosphorylation of specific Ser and Tyr residues in
the receptor large C-terminal domain (129).
Another mechanism that may participate in B2R
Fig. 2.
Kinin receptors and their signaling pathways. Schematic
representation of B
1
and B
2
receptors and the second messengers
released by their activation. PLC: phospholipase C; ER: endothelial
reticulum; DAG: diacylglycerol; IP3: inositol 1,4,5-triphosphate;
PLA
2
: phospholipase A
2
; NO: nitric oxyde; eNOS: endothelial NO
synthase.

The Kallikrein-Kinin System
19
desensitization involves BK-promoted transient associa-
tion of G
α
q
and G
α
i
with in caveolae (183). B2R acti-
vation leads to functional desensitization which is
associated with receptor phosphorylation
/
dephosphory-
lation and endocytosis
/
surface re-expression cycles
(184). Cys
324
in the cytoplasmic carboxyl terminus of
the human B2R appears to play a role in agonist-induced
internalization. It is true that B2R down-regulation is
observed in some forms of intense and chronic inflam-
mation, but the mechanism is unknown; proteases
present in the extracellular fluid have been shown to
destroy a form of recombinant B2R, thus providing a
possible mechanisms for inflammatory B2R down-
regulation (185).
The B1R differs from the B2R in that it is desensitized
only to a very limited degree and human B1R is not
phosphorylated to any significant degree either in the
absence or presence of agonist (186). The receptor
lacks any Ser and Tyr residues in the C-terminal tail.
This lack of regulation can contribute to the constitutive
activity of the receptor. Its agonist-induced translocation
to caveolae-related rafts without internalization has
been proposed (129, 187). This type of reversible
translocation may be of interest for a subset of the
signaling pathways activated by kinin receptors, as rafts
are rich in signaling molecules.
Part II: The Kallikrein-Kinin System: Pathophysiology
and Pharmacological Target
I- The Kinin Forming System in Plasma
1. Genetic defects
1.1 Defects of the contact system components
Genetic deficiencies have been reported for HK
(William trait), PKK (Fletcher trait), and factor XII
(Hageman trait) (188). These defects do not lead to
bleeding tendencies. Deficiencies of factor XII, forever
associated with thromboembolic events, suggest a
relationship between depressed factor XII-dependent
fibrinolysis and cardiovascular diseases. Quantitative
and qualitative defects of plasminogen have also been
associated with thromboembolism diseases (19, 189).
1.2 Defect in the control of the contact system: C1
inhibitor (C1INH)
1.2.1 Definition
Patients who present a genetic deficiency in C1INH
suffer from hereditary angioedema (HAE). HAE attacks
involve the activation of two pathways controlled by
this serpin: the classical complement and the contact
system pathways. The latter is responsible for the release
of vasoactive BK (190), which is probably the main
but not the sole mediator responsible for the increased
vascular permeability that results in angioedema (AE)
(191 – 194). The activation of other pathways could
also be involved in the pathogenesis of HAE. In fact,
Cugno et al. (1993) reported that generation of BK is
associated with activation of fibrinolysis during acute
attacks of HAE (51).
The prevalence of HAE is believed to be between
1
/
10,000 and 1
/
50,000 people worldwide (195). HAE
is traditionally described as Type I (HAE-I, 85% of
patients), which is characterized by a defective or
absence in gene production of C1INH and Type II
(HAE-II, 15% of patients), which is characterized by a
functionally impaired C1INH (196). C1INH deficiency
is heterogeneous at the gene level and is caused by
subtle changes affecting one or several nucleotides,
large deletions or duplications. These modifications
have been discussed recently by Agostoni (190). In
either case, HAE is associated with low functional
activity of C1INH, low levels of C4, and normal levels
of C3.
1.2.2 Pathophysiology
A murine model of HAE contributed to support the
hypothesis that BK mediates HAE. In this model, mice
heterozygous and homozygous for a gene coding for
C1INH demonstrated increased permeability and deple-
tion of HK. When treated with a specific plasma
kallikrein inhibitor or a B2R antagonist, the increased
vascular permeability was completely reversed (197).
Besides HAE, acquired forms of angioedema (AAE)
have been described. These AAE, characterized by
normal immunoreactive and functional C1INH levels,
are associated with drug therapy, such as with estrogens
and metallopeptidase inhibitors (ACE inhibitors (ACEi)
and vasopeptidases inhibitors (VPi), see below) (198).
They also occur during immunoproliferative and
autoimmune diseases. Finally, idiopathic forms of AAE
have also been reported (190).
1.2.3 Treatment of HAE
The purpose of this treatment is to inhibit the release
of vasoactive peptides among which is BK or to block
their proinflammatory effects (Fig. 3).
1.2.3.1 Serine proteases inhibitors
: C1INH and
aprotinin are two serpins used in the treatment of HAE
(199).
A) Aprotinin and aprotinin-like drugs
: Aprotinin is a
naturally occuring 58 aminoacid serpin isolated from
bovine lung that inhibits serine proteases with a parti-
cularly high affinity for plasma kallikrein and plasmin
(200). Inhibition of plasma kallikrein, which triggers the
release of BK during the contact system activation,

ME Moreau et al
20
could lead to a decreased release of BK during the HAE
attack. This inhibition of plasmin, but also of other
coagulation factors like protein C, is probably respon-
sible for the effectiveness of aprotinin in the treatment of
bleeding of different ethiologies (disseminated intra-
vascular coagulation, extracorporeal circulation during
cardiac surgery).
An aprotinin-like inhibitor isolated from human
urine, ulinastatin, is approved for intravenous therapy
in Japan (200). Nafamostat is a synthetic inhibitor of
kallikrein and coagulation factors enzymes (201, 202)
by working as an inverse substrate.
B) C1INH, Berinert
®
P
: Severe HAE-attacks are
currently treated by intravenous injection of pasteurized
C1INH purified from human blood plasma (pdC1INH;
Berinert
®
P) (203). This treatment is efficacious as it
inhibits the activation of the contact system that typi-
cally occurs during HEA attacks.
Recombinant human C1INH has also been developed
and is currently being tested in a clinical trial.
1.2.3.2 DX88
: DX88 is a synthetic kallikrein-inhibi-
tor, based on a recombinant Kunitz-domain (a serine
protease inhibitor domain) produced by a phage display
technology. In vivo, the drug effectively reverses the
increased vascular permeability in C1INH-deficient
mice at very low intravenous doses (193 – 197, 204).
As DX88 bypasses the C1INH pathway, it presents
also a potential interest in the treatment of AAE. Clinical
trials show that the drug was generally well tolerated and
improved the clinical symptoms of HAE within the first
4 h following the laryngeal attack.
1.2.3.3 Attenuated androgens: Danazol
®
, Stanozolol
®
:
Danazol and stanozolol are synthetic analogues of 17-
α
-
alkylated androgen that were shown to considerably
reduce the number HAE attacks when used for long-
term prophylaxis. As testosterone derivatives, they
maintain a residual hormonal activity whose clinical
relevance is dependent on the dose. High doses of
attenuated androgens (400 – 600 mg
/
day) correct the
biochemical defect of HAE, normalizing C1INH and
C4, usually leading to complete disappearance of AE
crises after a month of treatment (205).
The mechanism of action of attenuated androgens is
probably related to an increase in protein synthesis, as
an increase in C1INH plasma levels is observed (206),
but the mechanism has not yet been elucidated (196),
and the effect on metallopeptidases responsible for the
inactivation of kinins has not been documented.
1.2.3.4 Antifibrinolytic drugs: tranexamic acid
(Transamin
®
, Cyklokapron
®
, Exacyl
®
, Cyklo-f
®
)
:
Tran-
examic acid (4-(aminomethyl)cyclohexanecarboxylic acid)
is a synthetic lysine derivative that forms a reversible
complex with plasminogen at the lysine binding site and
thus prevents fibrin degradation by plasmin without any
effect on overall blood coagulation parameters. The
reduction in plasminogen binding to fibrin appears to
result in a decrease in the production of t-PA by endo-
thelial cells or an increase in the rate of its clearance
(207).
Tranexamic acid is presented in a variety of formula-
tions for oral or intravenous use. The oral bioavailability
has a rate of approximately 34% and therapeutic plasma
concentrations reach 5 – 10 mg
/
L (208). A greatest
efficacy against HAE was reported when the drug was
taken in a long-term prophylaxis (205, 207, 208).
1.2.3.5 B2R antagonists: HOE 140 (Icatibant
®
or
JE049
®
)
: Icatibant is currently tested in clinical trials
for treatment of HAE. A phase II proof-of-concept study
in HAE was concluded with positive clinical results.
High bioavailability (about 90%), combined with a low
variability and a maximal concentration reached after
about 30 min, has been demonstrated with a sub-
cutaneous formulation, which can be self-administrated
at a very early stage of an HAE attack. Icatibant has
also shown therapeutic benefit in refractory ascites in
liver cirrhosis as well as preclinical models for severe
burn injuries (209).
Fig. 3.
Pharmacological targets to modulate the kallikrein-kinin
activity. Tranexamic acid inhibits fibrinolysis and DX88 and C1INH
inhibit the serine activity of plasma kallikrein, although androgens
stimulate the synthesis of C1INH. B
1
and B
2
antagonists block the
activation of their respective receptors.