Файл: Методические указания Печатается по решению учебнометодической комиссии химического факультета Иркутского государственного.doc
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 22.11.2023
Просмотров: 59
Скачиваний: 2
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
СОДЕРЖАНИЕ
0,1 н раствор НСI.
0,1 н раствор СН3СООН.
0,1 н раствор NаОН.
Потенциометр ЛПМ-60М (или ЭВ-74).
Индикаторный электрод – стеклянный.
Электрод сравнения – хлорсеребряный.
Бюретка для титрования.
Электромагнитная мешалка со стержнем для перемешивания раствора.
Выполнение работы
1. Включить прибор и прогреть 30 мин.
2. Титрование соляной кислоты. В стаканчик для титрования поместить 5 мл 0,1 н раствора НСl (раствор отобрать пипеткой), разбавить дистиллированной водой, аккуратно опустить в раствор электроды, стержень-мешалку, включить магнитную мешалку и отрегулировать скорость перемешивания (скорость должна быть такой, чтобы при перемешивании отсутствовала воронка между электродами).
Бюретку для титрования заполнить титрантом 0,1 н раствором NаОН, конец бюретки опустить в стаканчик для титрования так, чтобы он не касался раствора, стенок стакана и электродов.
По нижней шкале потенциометра сделать первый отсчет, затем из бюретки прилить 0,5 мл 0,1 н раствор NаОН, раствор перемешать, сделать отсчет по шкале Е, мВ. Все результаты (первый и далее) внести в табл. 3. Титрование вести через 0,5 мл до того значения потенциала, пока оно не станет либо постоянным, либо изменение будет незначительным.
Таблица 3
Результаты потенциометрического титрования
| Количество анализируемой кислоты, мл | Количество прилитой 0,1 н щелочи, мл | Показания прибора, в мВ | Для смеси | ||||
| НСI | СН3СООН | НСI+ СН3 СООН | Е | V | E/V | ||
| НСl, 5 | 0,0 | | | | | | |
| | 0,5 | | | | | | |
| | 1,0 | | | | | | |
| | 1,5 | | | | | | |
| | …… | | | | | | |
3. Титрование уксусной кислоты. 5 мл 0,1 н раствора уксусной кислоты поместить в стаканчик, разбавить дистиллированной водой до метки, опустить стержень-мешалку, заполнить бюретку едким натром и провести титрование как и в предыдущем случае. Результаты внести в табл. 3.
4. Титрование смеси кислот. В стаканчик поместить указанный преподавателем объем анализируемой смеси кислот, добавить 20 мл ацетона, разбавить до 50 мл водой. Подготовить к титрованию бюретку и оттитровать смесь кислот 0,1 н раствором NаОН, прибавляя последний по 0,5 мл. Данные свести в табл. 3.
Следует помнить, что электроды после титрования вынимают из раствора, предварительно переведя в нулевое положение переключатель «Род работы». Затем их ополаскивают водой и осушают фильтровальной бумагой. В конце работы чистые электроды помещают в специальные пробирки для хранения.
5. Построить интегральные кривые потенциометрического титрования растворов на миллиметровой бумаге в координатах: Е, мВ – V, мл (рис. 21, а) и дифференциальную кривую только для смеси кислот в координатах ΔE/ΔV = f(V) (рис. 21, б).
На кривых титрования найти точку эквмвалентности, рассчитать количество кислот через титр по определяемому веществу. Сопоставить полученные результаты, сделать заключение о возможности дифференцированного определения соляной и уксусной кислот в их смеси.
Расчет
Содержание соляной (m1) и уксусной (m2) кислот в смеси вычисляют по формулам:
m1 = T0,1MNaOH/HCl · V1
m2 = T0,1MNaOH/CH3COOH · (V2 – V1)
где V1 – объем NaOH, найденный из кривой титрования по первой ступени;
V2 – объем NaOH, найденный из кривой титрования по второй ступени.
1 2 3 4
ЛАБОРАТОРНАЯ РАБОТА 6
Раздельное определение соляной и серной кислот в их смеси
потенциометрическим титрованием в неводной среде
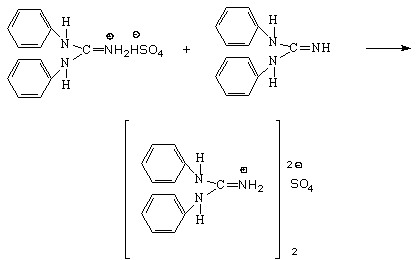
Раздельное определение соляной и серной кислот в их смеси основано на прямом дифференцированном титровании стандартным спиртовым раствором дифенилгуанидина (ДФГ) в среде ацетона. ДФГ представляет собой органическое основание. В этих условиях кривая потенциометрического титрования смеси кислот характеризуется двумя скачками. Первый скачок соответствует совместному титрованию обеих кислот по первой ступени диссоциации, второй – титрованию серной кислоты по второй ступени.
Если в растворе присутствует только серная кислота, то на титрование ионов водорода, образующихся в процессе диссоциации по обеим ступеням, расходуются равные объемы стандартного раствора основания.
Титрование кислот раствором ДФГ протекает по следующим уравнениям:
Первая ступень
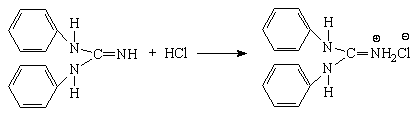
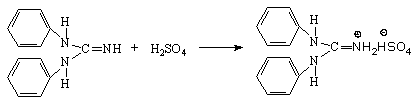
Вторая ступень (только для серной кислоты)
Реагенты и аппаратура
0,1 н спиртовый раствор дифенилгуанидина.
0,1 н ацетоновый раствор НСI.
0,1 н ацетоновый раствор Н2SO4.
Растворитель – ацетон.
Потенциометр ЛПМ-60М (или ЭВ-74).
Два электрода – индикаторный стеклянный электрод и сравнения – хлорсеребряный, закрепленные в штативе.
Бюретка для титрования.
Электромагнитная мешалка со стержнем для перемешивания раствора.
Выполнение работы
1. Титрование серной кислоты. В стаканчик для титрования помещают 3 мл серной кислоты, затем приливают 20 мл ацетона, помещают стержень-мешалку. Опускают электроды и включают магнитную мешалку.
Бюретку заполняют 0,1 М спиртовым раствором ДФГ. Титрант добавляют порциями по 0,2 мл. После второй точки эквивалентности титрование продолжают до тех пор, пока не будут изменяться значения потенциала. Во время титрования серной кислоты может наблюдаться выпадение осадка сульфата дифенилгуанидиния, хорошо растворимого в воде. Для растворения осадка в ячейку по каплям добавляют дистиллированную воду до полного осветления раствора и продолжают титрование. После окончания титрования переключатель «род работы» переводят в нулевое положение. Электроды вынимают из раствора, ополаскивают водой и осушают фильтровальной бумагой.
2. Титрование соляной кислоты. 3 мл соляной кислоты помещают в стаканчик для титрования, приливают 20 мл ацетона, бюретку заполняют раствором дифенилгуанидина и проводят титрование, как описано для серной кислоты. Хлорид дифенилгуанидиния растворим в среде ацетона.
3. Титрование смеси кислот. В ячейку для титрования помещают 1 мл соляной и 2 мл серной кислот, добавляют 20 мл ацетона и выполняют потенциометрическое титрование 0,1 М раствором дифенилгуанидина, приливая титрант по 0,2 мл. Результаты трех титрований занести в табл. 4.
4. Построить кривые титрования в координатах: E = f(V).
Таблица 4
Результаты потенциометрического титрования
| V анализируемой кислоты, мл | V 0,1 н NaOH, мл | Показания прибора E, мВ | m (кислот), г/мл | ||||
| НСI | Н2SO4 | НСI+Н2SO4 | кислота | индивидуально | в смеси | ||
| НСl 3 | 0,0 | | | | Н2SO4 | – | – |
| | 0,2 | | | | |||
| | 0,4 | | | | НСI | – | – |
| | 0,6 | | | | |||
| | …… | | | | |||
Расчет
Содержание серной (m1) и соляной (m2) кислот в смеси вычисляют по формулам:
m1 = T0,1MДФГ/H2SO4 · 2 (V2 – V1)
m2 = T0,1MДФГ/Cl · (V1 – (V2 – V1))
где V1 – объем ДФГ, найденный из кривой титрования по первой ступени;
V2 – объем ДФГ, найденный из кривой титрования по второй ступени.
4. КОНДУКТОМЕТРИЯ
Методы анализа, связанные с измерением электропроводности растворов, называются кондуктометрическими.
Электропроводность – это способность вещества проводить электрический ток под действием внешнего электрического поля. Величина электропроводности W выражается через сопротивление раствора R и измеряется в обратных омах или Сименсах, [Ом-1] = [См]:
При этом сопротивление раствора прямо пропорционально удельному сопротивлению ρ проводника и отношению длины проводника L к площади поперечного сечения S:
Величина, обратная удельному сопротивлению, называется удельной электропроводностью χ, и характеризует электропроводность 1 см3 раствора:
Единицы измерения удельной электропроводности – [См/см].
Связь между электропроводностью W и удельной электропроводностью χ:
Различают молярную (μ) и эквивалентную (λ) электропроводности.
Молярная электропроводность (μ) – это удельная электропроводность 1 грамм-моля (η) вещества в 1 см3 раствора:
Единицы измерения – [См·см2/моль].
Эквивалентная электропроводность (λ) – это удельная электропроводность 1 грамм-эквивалента вещества в 1 см3 раствора:
Единицы измерения – [См·см2/г-экв.].
Электропроводность растворов электролитов зависит от нескольких факторов: концентрации, температуры, природы электролита и растворителя.
Влияние концентрации на электропроводность растворов электролитов. В растворах сильных электролитов большое влияние оказывают межионные взаимодействия. С повышением концентрации электропроводность растворов увеличивается за счет увеличения переносчиков тока (ионов). При дальнейшем увеличении концентрации она проходит через максимум и начинает уменьшаться, так как в этих условиях ионы начинают сильнее взаимодействовать друг с другом, и образуются электронейтральные молекулы, не проводящие тока (рис. 23). В свою очередь, эквивалентная электропроводность растворов сильных электролитов возрастает с уменьшением концентрации (рис. 24, кривая 1) и выражается уравнением:
где λ0 – предельная эквивалентная электропроводность при бесконечном разбавлении; а – константа.
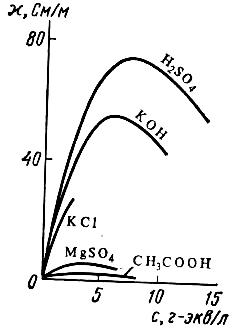
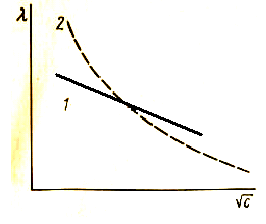
| Рис. 23. Зависимость удельной электропроводности различных электролитов от концентрации | Рис. 24. Эквивалентная электропроводность электролитов: 1 – сильного; 2 – слабого |
По теории Дебая–Хюккеля–Онзагера уменьшение эквивалентной электропроводности с увеличением концентрации объясняется эффектами электрофоретического и релаксационного торможения. Именно константа а из вышеуказанного уравнения учитывает эти эффекты.
Электрофоретический эффект вызывается тем, что центральный ион под действием электрического поля движется в одном направлении, а ионная атмосфера – в другом и тем самым тормозит движение иона.
Релаксационный эффект обусловлен процессами разрушения и формирования ионной атмосферы при движении иона.
В случае слабых электролитов большое влияние на электропроводность оказывает степень диссоциации. При разбавлении растворов слабых электролитов эквивалентная электропроводность возрастает, так как увеличивается степень их диссоциации, а также сказывается влияние двух вышеупомянутых эффектов (рис. 24, кривая 2).
Влияние температуры на электропроводность. С повышением температуры электропроводность увеличивается, так как уменьшается вязкость раствора. И, следовательно, повышается подвижность ионов. Зависимость удельной электропроводности от температуры выражается степенным рядом:
χt = χ0(1+ αt + βt2+…)
где χ0 – удельная электропроводность при 25 ºС; α и β – коэффициенты, зависящие от природы электролита и концентрации; t – температура раствора.
Влияние природы электролита и растворителя на электропроводность. Ионы обладают различной подвижностью и, следовательно, проводимостью. Самые большие подвижности у ионов водорода Н+ (349,8) и гидроксид-иона ОН- (198,3). Это объясняется «эстафетным» механизмом передвижения. Влияние природы электролита на электропроводность выражается законом Кольрауша:
где U+ и V- – подвижности катиона и аниона соответственно.
Растворитель также оказывает влияние на электропроводность, поскольку обладает определенной вязкостью и диэлектрической проницаемостью. В растворителях с низкой диэлектрической проницаемостью наблюдаются процессы ассоциации ионов и, следовательно, понижение проводимости.
Измерение электропроводности (или сопротивления) растворов осуществляют компенсационным методом с помощью моста Кольрауша (рис. 25).
Д
Рис. 25. Схема моста Кольрауша
1 – нуль-индикатор; 2 – генератор переменного тока; Rм – стандартное сопротивление; Rx – ячейка с раствором; аб – реохорд; ав и вб – плечи реохорда
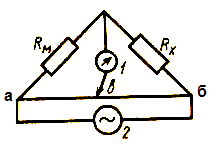 ля определения значения Rхизменяют положение передвижного контакта в до тех пор, пока нуль-индикатор 1 не будет показывать отсутствие тока в цепи или же его минимальное значение. Сопротивление в ячейке можно рассчитать:
ля определения значения Rхизменяют положение передвижного контакта в до тех пор, пока нуль-индикатор 1 не будет показывать отсутствие тока в цепи или же его минимальное значение. Сопротивление в ячейке можно рассчитать: где lвб и lав – длины соответствующих плеч.
Константа сосуда. Экспериментально измеряемая величина сопротивления раствора зависит не только от размера электродов и расстояния между ними, а также от формы, их взаимного расположения, объема раствора и других факторов, не всегда поддающихся точному учету, так как токопроводящим является не только объем раствора, который заключен между электродами. Поэтому для определения величины электропроводности раствора используют константу сосуда (С), которая представляет собой соотношение:
Для определения константы сосуда применяют стандартные растворы хлорида калия, величина удельной электропроводности которых при различных температурах установлена с большой точностью (табл. 5).
Методы кондуктометрии подразделяются на прямые и косвенные (кондуктометрическое титрование). Методы прямой кондуктометрии используются значительно реже косвенных, так как значения подвижностей ионов достаточно близки и в этом случае можно получить информацию лишь об общей концентрации ионов в растворе. С помощью прямой кондуктометрии можно определять некоторые физико-химические свойства и характеристики веществ: константы диссоциации, растворимость труднорастворимых веществ, можно изучать кинетику некоторых реакций. В аналитических целях прямую кондуктометрию используют, главным образом, при контроле качества дистиллированной воды, при анализе минеральных вод.
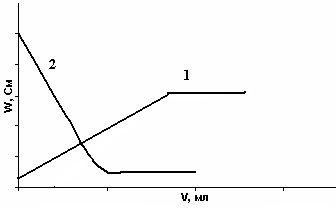
Большее распространение получили методы косвенной кондуктометрии – кондуктометрическое титрование. В методе кондуктометрического титрования используют реакции нейтрализации, осаждения, комплексообразования и реже окисления-восстановления. Кривые титрования строятся в координатах R = f(V), W = f(V),
χ = f(V). Точка эквивалентности находится путем продления прямолинейных участков до их пересечения. Рассмотрим кривые кондуктометрического титрования на примере реакций нейтрализации.
Метод нейтрализации
Кривые титрования представлены на рис. 26.
-
Титрование сильной кислоты сильным основанием:
HCl + NaOH → NaCl + H2O
До точки эквивалентности электропроводность раствора уменьшается вследствие того, что концентрация очень подвижных ионов водорода уменьшается. После точки эквивалентности, когда в раствор вводится избыток сильного электролита NaОН, содержащего подвижные гидроксид-ионы, электропроводность начинает возрастать, но не так резко, так как подвижность ОН- -ионов ниже, чем у ионов водорода (кривая 1).
-
Титрование слабой кислоты сильным основанием:
СН3СООН + NaOH → СН3СООNa + H2O
Д
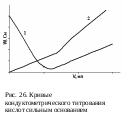 о точки эквивалентности наблюдается небольшой рост электропроводности несмотря на то, что происходит связывание очень подвижных ионов водорода. Это объясняется тем, что уксусная кислота достаточно слабо диссоциирована и свободных ионов водорода в растворе мало. В ходе реакции образуется ацетат натрия – сильный электролит, концентрация которого возрастает, что обуславливает увеличение электропроводности. После точки эквивалентности происходит еще больший рост электропроводности, так как в раствор вводится избыток сильного электролита NaОН, содержащего подвижные гидроксид-ионы (рис. 26, кривая 2). В этом случае точка эквивалентности выражена слабо вследствие гидролиза образующегося ацетата натрия.
о точки эквивалентности наблюдается небольшой рост электропроводности несмотря на то, что происходит связывание очень подвижных ионов водорода. Это объясняется тем, что уксусная кислота достаточно слабо диссоциирована и свободных ионов водорода в растворе мало. В ходе реакции образуется ацетат натрия – сильный электролит, концентрация которого возрастает, что обуславливает увеличение электропроводности. После точки эквивалентности происходит еще больший рост электропроводности, так как в раствор вводится избыток сильного электролита NaОН, содержащего подвижные гидроксид-ионы (рис. 26, кривая 2). В этом случае точка эквивалентности выражена слабо вследствие гидролиза образующегося ацетата натрия.-
Титрование слабой кислоты слабым основанием:
СН3СООН + NН4OH → СН3СООNН4 + H2O
До точки эквивалентности имеют место те же закономерности, что и в предыдущем случае. После точки эквивалентности практически не происходит изменения электропроводности, так как в раствор вводится избыток слабого электролита NН4ОН (рис. 27, кривая 1). В этом случае точка эквивалентности выражена менее четко, вследствие гидролиза образующегося ацетата аммония.
-
Титрование сильной кислоты слабым основанием:
НСl + NН4OH → NН4Cl + H2O
Д
Рис. 27. Кривые кондуктометрического титрования кислот слабым основанием
о точки эквивалентности электропроводность уменьшается вследствие того, что очень подвижные ионы водорода соляной кислоты связываются в молекулы воды. После точки эквивалентности практически не происходит изменения электропроводности, так как в раствор вводится избыток слабого электролита NН4ОН (рис. 27, кривая 2). В этом случае точка эквивалентности выражена слабо вследствие гидролиза образующегося хлорида аммония.
-
Титрование смеси сильной и слабой кислот сильным основанием:
H
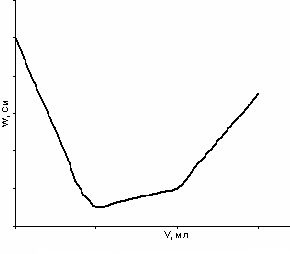
Cl + СН3СООН + NaOH → NaCl + СН3СООNa + H2O
К
Рис. 28. Кривая кондуктометрического титрования смеси кислот щелочью
ак и в классическом титровании, раздельно можно оттитровать две кислоты при совместном присутствии, если их константы диссоциации отличаются не менее чем в 104. Кривая титрования представлена на рис. 28. Первой титруется соляная (сильная) кислота, а затем уксусная. Падение электропроводности обусловлено уменьшением концентрации ионов водорода. После первой точки эквивалентности небольшой рост электропроводности объясняется образованием сильного электролита – ацетата натрия. И дальнейшее увеличение электропроводности после второй точки эквивалентности связано с введением в раствор избытка щелочи.
-
Титрование смеси сильной и слабой кислот слабым основанием:
HCl + СН3СООН + NН4OH → NaCl + СН3СОО NН4 + H2O
К
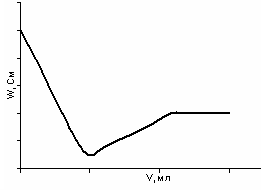
ривая титрования изображена на рис. 29. До первой точки эквивалентности падение электропроводности связано с уменьшением концентрации ионов водорода. После первой точки эквивалентности небольшой рост электропроводности объясняется образованием сильного электролита – ацетата аммония. После второй точки эквивалентности практически не происходит изменения электропроводности, так как в раствор вводится избыток слабого электролита NН4ОН.
-
Т
Рис. 29. Кривая кондуктометрического титрования смеси кислот слабым основанием
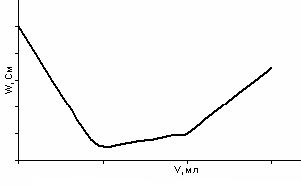
итрование двухосновной кислоты сильным основанием. По двум ступеням можно оттитровать те двухосновные кислоты, которые хорошо диссоциируют по первой ступени (рКI < 2,5), но слабо диссоциируют по второй ступени (рКII = 6–10). Например, хромовая кислота титруется щелочью по двум ступеням (рКI = 1; рКII = 6,5):
H2CrO4 + NaOH → NaHCrO4 + H2O
NaHCrO4 + NaOH → Na2CrO4 + H2O
До первой точки эквивалентности хромовая кислота титруется как сильная, после этого идет ее титрование как слабой кислоты. Кривая титрования изображена на рис. 30.
П
Рис. 30. Кривая кондуктометрического титрования хромовой кислоты щелочью
ри использовании реакций осаждения вид кривой зависит от того, какой ион замещается: более подвижный на менее подвижный (наблюдается падение электропроводности) или же наоборот (возрастание электропроводности). Если в растворе присутствует несколько ионов, то первым осаждается тот, который образует менее растворимый осадок. Кроме того, значения произведения растворимости образующихся осадков должны отличаться не менее чем на 106. При использовании реакций осаждения необходимо помнить, что повышение растворимости осадка приводит к тому, что точка эквивалентности на кривой титрования будет размыта.
В случае реакций комплексообразования (рис. 31) чаще всего в качестве титранта используют комплексон III (Трилон Б). При взаимодействии катионов металла с Трилоном Б увеличивается концентрация ионов водорода и наблюдается увеличение электропроводности:
Me2+ + H2Y2- → MeY2- + 2H+
После точки эквивалентности электропроводность начинает падать, так как выделившиеся ионы водорода связываются анионом Трилона Б:
H
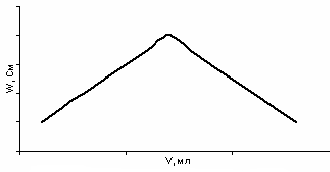
Рис. 31. Кривая кондуктометрического титрования Трилоном Б
+ + H2Y2- → H3Y-
Если же титрование проводить в буферных растворах, то после точки эквивалентности наблюдается увеличение электропроводности вследствие увеличения концентрации титранта. Выделяющиеся ионы водорода связываются буферной системой и не вносят значимого вклада в электропроводность.
ЛАБОРАТОРНАЯ РАБОТА 7
Определение соляной кислоты в растворе методом
кондуктометрического титрования
Цель работы: определить содержание кислоты в анализируемой пробе.
Реагенты и аппаратура
Проба НСl.
0,01 н раствор NaOH.
0,1н раствор KCl.
Термометр.
Ячейка с платиновыми электродами.
RLC-мост.
Бюретка для титрования.
Электромагнитная мешалка со стержнем для перемешивания раствора.
Методика работы
1. Определение константы сосуда. В химический стакан наливают 0,1 н раствор KCl так, чтобы платиновые электроды были закрыты. Измеряют сопротивление этого раствора. Константа сосуда вычисляется по формуле:
C = R ,
где С – константа сосуда;
– удельная электропроводность 0,1 н раствора KCl (см. табл. 5);
R – сопротивление раствора 0,1 н KCl, измеренное на приборе.
2. Титрование соляной кислоты. В стакан вместо раствора KCl наливают 10 мл 0,01 н раствора соляной кислоты, разбавляют водой так, чтобы электроды были полностью покрыты раствором и титруют 0,01 н раствором едкого натра, прибавляя его по 0,5 мл. Раствор перемешивают электромагнитной мешалкой и измеряют сопротивление. Титрование прекращают после того, как сопротивление уменьшается вследствие введения избытка щелочи. По полученным данным строят график зависимости сопротивления R (ось ординат) от количества мл титрованного раствора щелочи (ось абсцисс). Вычисляют W и , строят графики зависимости W, (ось ординат) от объема раствора щелочи (ось абсцисс). Определяют точку эквивалентности титрования кислоты щелочью и рассчитывают содержание кислоты в пробе, в граммах.
m = T0,1MNaOH/HCl · V0,1MNaOH
Таблица5
Удельная электропроводность раствора KCl
| Температура раствора | 1 н 103 | 0,1 н 103 | Температура раствора | 1 н 103 | 0,1 н 103 |
| 0 | 6,541 | 7,15 | 19 | 10,14 | 11,43 |
| 5 | 7,414 | 8,22 | 20 | 10,207 | 11,67 |
| 10 | 8,310 | 10,33 | 21 | 10,40 | 11,97 |
| 15 | 9,252 | 10,66 | 22 | 10,95 | 12,15 |
| 16 | 9,441 | 10,72 | 23 | 10,789 | 12,39 |
| 17 | 9,631 | 10,95 | 24 | 10,984 | 12,64 |
| 18 | 9,822 | 11,619 | 25 | 11,180 | 12,88 |
ЛАБОРАТОРНАЯ РАБОТА 8
Кондуктометрическое определение серной кислоты
и сульфата меди в смеси
Цель работы: определить содержание серной кислоты и сульфата меди в граммах в анализируемом растворе.
Анализируемая проба представляет собой смесь серной кислоты и сульфата меди. Титруем смесь раствором едкого натра. Сначала титруется серная кислота:
H2SO4 + 2NaOH Na2SO4 + 2H2O
Электропроводность раствора быстро уменьшается, так как в результате реакции сильно подвижный ион водорода (UH+ = 349,7) замещается на менее подвижный ион натрия (UNa+= 50). После достижения первой точки эквивалентности при дальнейшем прибавлении раствора щелочи электропроводность смеси практически изменяться не будет, так как в растворе ионы меди (UCu2+ = 56) замещаются ионами натрия, подвижности которых близки. Уравнение реакции:
CuSO4 + 2NaOH Cu(OH)2 + Na2SO4
Как только закончится реакция осаждения гидроксида меди, дальнейшее прибавление едкого натра приведет к значительному увеличению электропроводности раствора вследствие появления в растворе избытка ионов ОН-.
Реагенты и аппаратура
Пробы
смеси Н2SO4 + CuSO4.
0,02 н раствор NaOH.
0,1 н раствор KCl.
Термометр.
Ячейка с платиновыми электродами.
Кондуктометр ОК 1021.
Бюретка для титрования.
Электромагнитная мешалка со стержнем для перемешивания раствора
Выполнение работы
1. Определение константы сосуда. Определение константы сосуда проводят точно так же, как и в лабораторной работе 7 (см. с. 50).
2. Титрование смеси серной кислоты и сульфата меди. В стакан наливают 10 мл пробы, разбавляют водой так, чтобы электроды были полностью покрыты раствором и титруют 0,02 н раствором едкого натра, приливая его по 0,5 мл. Титрование прекращают после того, как электропроводность раствора значительно возрастет вследствие введения в раствор избытка щелочи. По полученным данным строят график зависимости электропроводности (ось ординат) от количества мл титрованного раствора щелочи (ось абсцисс). Определяют точки эквивалентности титрования кислоты и сульфата меди, после чего рассчитывают содержание каждого компонента в смеси.
m1 = T0,02MNaOH/H2SO4 · V1
m2 = T0,02MNaOH/ CuSO4 · (V2 – V1)
ЛИТЕРАТУРА
-
Васильев В. П. Аналитическая химия / В. П. Васильев. – М. : Высш. шк., 1989. – Т. 2. -
Виноградова Е. H. Методы полярографического и амперометрического анализа / Е. H. Виноградова, З. A. Галлай, З. M. Финогенова. – M., 1963. -
Гейровский Я. Основы полярографии / Я. Гейровский, Я. Кута. – М. : Мир, 1965. -
Золотов Ю. А. Основы аналитической химии / Ю. А. Золотов. – М. : Высш. шк., 2004. – Т. 2. -
Коваленко П. Н. Физико-химические методы анализа / П. Н. Коваленко, Е. Н. Багдасаров. – М. : Мир,1975. -
Лопатин Б. А. Теоретические основы электрохимических методов анализа / Б. А. Лопатин. – М. : Высш. шк., 1975. -
Ляликов Ю. C. Физико-химические методы анализа / Ю. С. Ляликов. – M. ; Л., 1964. -
Плэмбэк Дж. Электрохимические методы анализа. Основы теории и применение / Дж. Плэмбэк. – М. : Мир, 1985.
Учебное издание
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Часть 2
Электрохимические методы анализа
Методические указания
Составители:
Апрелкова Нина Федоровна,
Королева Галина Николаевна,
Матвеев Дмитрий Александрович,
Недвецкая Галина Борисовна
Подготовлено к печати: Г. А. Никифорова
Подписано в печать 30.01.2009. Формат 60х84 1/16.
Печать трафаретная. Усл. печ. л. 3,0. Уч.-изд. л. 1,9.
Тираж 100 экз. Поз. 3. Заказ 10.
Издательство Иркутского государственного университета
664003, Иркутск, бульвар Гагарина, 36; тел. (3952) 24-14-36