Файл: Методические указания Печатается по решению учебнометодической комиссии химического факультета Иркутского государственного.doc
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 22.11.2023
Просмотров: 58
Скачиваний: 2
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
СОДЕРЖАНИЕ
Еразл. – Еравн. = η
Тогда условием протекания непрерывного электролиза является:
Еразл. = (Еанода + ηанода) – (Екатода + ηкатода) + iR,
где iR – омическое сопротивление, создаваемое самой электролизной ячейкой.
Перенапряжение зависит от ряда факторов:
- величины плотности тока: чем больше плотность тока, тем больше перенапряжение;
- материала электрода;
- температуры.
При протекании электролиза в кислых средах наблюдается выделение газообразных водорода и (или) кислорода. При этом на электродах возникают соответствующие газовые полуэлементы. В связи с этим вводится понятие «перенапряжение водорода» или «перенапряжение любого другого газа». Значение перенапряжения водорода на платинированной платине равно 0 В (нулю вольт), в то время как на гладкой платине -0,07 В. В зависимости от природы металла перенапряжение водорода принимает различные значения (табл. 1).
Таблица 1
Значения перенапряжения водорода на различных металлах
при i = 0,01 А/см2
| Металл катода | Платинированная Pt | Гладкая Pt | Au | Fe | Cu | Zn | Hg |
| η, В | 0 | -0,07 | -0,39 | -0,56 | -0,58 | -0,75 | -1,04 |
Перенапряжение водорода на ртути – одно из самых высоких, что имеет практическое значение.
1.5. Разновидности электролиза
Существует три разновидности электролиза: ускоренный, электролиз с ртутным катодом, внутренний электролиз.
-
Ускоренный электролиз. Процесс выделения металла на катоде включает две стадии: диффузию ионов к катоду и осаждение металла на нем. Для ускорения электролиза необходимо влиять на эти две стадии.
Процесс диффузии ускоряют:
а) концентрация анализируемого раствора;
б) температура;
в) перемешивание.
Процесс осаждения зависит от:
а) вид иона металла: восстановление комплексного иона протекает медленнее, чем свободного;
б) плотности тока: чем выше плотность тока, тем больше металла осаждается в единицу времени. Однако при больших
плотностях тока осадок получается рыхлым, что приводит к большим потерям. Чаще всего плотность тока составляет 0,001–0,01 А/см2.
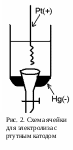
2. Электролиз с ртутным катодом. Вследствие высокого перенапряжения водорода на ртути и способности ее к образованию амальгам, обладающих меньшими окислительными потенциалами, чем сами выделяющиеся при электролизе металлы, электролиз с применением ртутного катода дает возможность проводить ряд разделений, имеющих большое практическое значение. Схема установки для электролиза с ртутным катодом изображена на рис. 2.
Все металлы по способности образовывать амальгамы делят на 4 группы (табл. 2).
Таблица 2
Группы металлов по способности давать амальгамы
| Содержание металла в ртути | |||
| I группа меньше 0,001 % | II группа 0,001–0,1 % | III группа 0,1–1 % | IV группа 1 % |
| Be, Fe, Ni | Cu, Al, Li | Na, Ba, Mg, Sr | Zn, Tl, Cd,Cs |
Железо, несмотря на столь низкую способность растворяться в ртути, образует коллоидный раствор с концентрацией до 0,8 %. Те металлы, которые не растворяются в ртути, при восстановлении находятся на поверхности ртути в виде шлама. Количественное определение выделившегося металла сложно провести, поскольку необходимо каждый раз взвешивать ртутный катод, что крайне неудобно.
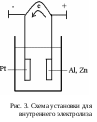
3. Внутренний электролиз. При проведении внутреннего электролиза не требуется наличие источника внешней ЭДС. Для протекания электролиза необходимо, чтобы анализируемый раствор с погруженными в него электродами представлял собой гальванический элемент, дающий собственный ток. Установка для проведения внутреннего электролиза представлена на рис. 3. Если в стакан поместить раствор соли меди и погрузить два электрода, один из которых платиновый (катод), другой цинковый (анод), то возникает гальванический элемент. При этом цинковый анод будет растворяться, отдавая электроны:
Z
 n0 – 2e → Zn2+
n0 – 2e → Zn2+Освободившиеся при этом электроны переходят на платину, где протекает процесс восстановления ионов меди:
Cu2+ + 2e → Cu0
Таким образом, в качестве анода должен использоваться металл, обладающий более отрицательным окислительно-восстановительным потенциалом, чем осаждающийся на катоде.
О
 собенностью внутреннего электролиза является то, что образующийся гальванический элемент дает равномерный слабый ток. Это позволяет получать на катоде плотный осадок, при этом электролизу подвергают растворы с малой концентрацией. Кроме того, единственным процессом на аноде является растворение самого анода.
собенностью внутреннего электролиза является то, что образующийся гальванический элемент дает равномерный слабый ток. Это позволяет получать на катоде плотный осадок, при этом электролизу подвергают растворы с малой концентрацией. Кроме того, единственным процессом на аноде является растворение самого анода.Главной опасностью внутреннего электролиза является цементация. Цементация – разряжение части определяемых ионов на аноде. Причины цементации: высокие концентрации, грязная поверхность анода, плохие контакты. Для предотвращения этого явления необходимо: зачищать контакты, использовать чистые металлы в качестве анода, разделять анодное и катодное пространства с помощью диафрагмы.
ЛАБОРАТОРНАЯ РАБОТА 1
Электровесовое определение меди и свинца
при совместном присутствии
Цель работы: определить процентное содержание меди и свинца в бронзе.
Теоретическое обоснование работы: свинец количественно выделяется на аноде в виде PbO2 из азотнокислых растворов. Ионы же меди в этих условиях всегда осаждаются на поверхности катода. Поэтому происходит одновременное выделение этих металлов из растворов. При растворении бронзы в азотной кислоте олово, если оно присутствует в данной пробе, окисляясь, выделяется в виде -оловянной кислоты:
Sn + 4 HNO3 + H2O 3 H2SnO3 + 4 NO
Другие металлы, входящие в состав бронзы, при действии азотной кислоты образуют растворимые нитраты, например:
Cu + 4 HNO3 Cu(NO3)2 + 2 NO2 + 2H2O
Pb + 4 HNO3 Pb(NO3)2 + 2 NO2 + 2H2O
Осадок H2SnO3 отделяют, и раствор подвергают электролизу.
При этом на электродах протекают следующие процессы:
Катод: Cu2+ + 2e Cu0
Анод: Pb2+ – 2e → Pb4+, а затем Pb4+ + 2H2O → PbO2 + 4H+
Реагенты и аппаратура
Проба бронзы.
HNO3 (1:1).
15%-ный растворNH4NO3.
H2SO4 (1:1).
10%-ный раствор NH4OH.
Спирт этиловый.
Диэтиловый эфир.
HNO3 концентрированная.
Платиновые электроды – 2 шт.
Аккумулятор на 4 В.
Методика выполнения работы: навеску 0,1 г бронзы помещают в стакан и растворяют в азотной кислоте (1:1). Кислоту приливают постепенно, порциями по 2–3 мл. Растворение ведут при нагревании (под тягой) на плитке. К полученному раствору добавляют 100 мл горячей дистиллированной воды, 20 мл 15%-го раствора нитрата аммония и кипятят 10–15 мин. Затем раствор упаривают до объема 100–120 мл и добавляют к нему 2,5 мл азотной кислоты 1:1.
В полученный раствор опускают электроды, предварительно взвешенные на аналитических весах. Электролиз ведут при напряжении 2,2–2,4 В и силе тока 1,8–3,0 А. Схема установки электролиза представлена на рис. 4.
Ч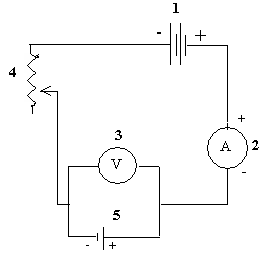 ерез 30–35 мин добавляют 2 мл серной кислоты (1:1) и, не прекращая электролиза, частично нейтрализуют раствор 2,5–3,0 мл 10%-го раствора NH4OH. Продолжают электролиз еще в течение 20–30 мин. Затем приливают в электролизер такое количество воды, чтобы уровень жидкости повысился на 1–1,5 см и проверяют выделение меди на свежей поверхности электрода. Е
ерез 30–35 мин добавляют 2 мл серной кислоты (1:1) и, не прекращая электролиза, частично нейтрализуют раствор 2,5–3,0 мл 10%-го раствора NH4OH. Продолжают электролиз еще в течение 20–30 мин. Затем приливают в электролизер такое количество воды, чтобы уровень жидкости повысился на 1–1,5 см и проверяют выделение меди на свежей поверхности электрода. Е
Рис. 4. Схема установки
для электролиза:
1 – аккумулятор на 4 В;
2 – амперметр;
3 – вольтметр;
4 – реостат;
5 – установка для электролиза с платиновыми электродами.
сли медь не выделяется из раствора, то, не прерывая тока, убирают стакан с раствором и промывают электроды, подставляя стакан с дистиллированной водой. После этого размыкают цепь, снимают электроды и промывают их спиртом, эфиром, высушивают над электроплиткой и взвешивают.
По окончании работы установку разбирают, электроды промывают: катод в концентрированной азотной кислоте, анод – в концентрированной азотной кислоте с добавлением 0,1 н щавелевой кислоты, затем в воде, спирте, эфире. После промывания электроды высушивают, взвешивают и сдают лаборанту.
Рассчитывают процентное содержание меди и свинца в бронзе.
ЛАБОРАТОРНАЯ РАБОТА 2
Определение меди в дюралюминии методом внутреннего электролиза в дюралюминии
Цель работы: определить процентное содержание меди в сплаве дюралюминия.
Реагенты и аппаратура
Проба дюралюминия.
20%-ный раствор NaOH.
2н раствор H2SO4.
HNO3 концентрированная.
Спирт этиловый.
Диэтиловый эфир.
Платиновый электрод – 1 шт.
Алюминиевый электрод – 1 шт.
Методика выполнения работы: в небольшом химическом стакане растворяют 0,1 г сплава дюралюминия в возможно малом количестве 20%-го едкого натра при нагревании (на плитке под тягой). Алюминий переходит в раствор, а медь, железо, марганец остаются в осадке:
2Al + 2NaOH + 6H2O → 2Na[Al(OH)4] + 3H2↑
Осадок растворяют при охлаждении в концентрированной азотной кислоте, которую добавляют по каплям (не более 1,5 мл):
Na[Al(OH)4] + 4 HNO3 → Al(NO3)3 + NaNO3 + 4H2O
Cu + 4HNO3 → Cu(NO3)2 + 2NO2↑ + 2H2O
После нагревания осадка и растворения его, к исследуемому раствору добавляют 100 мл 2 н серной кислоты, доводят дистиллированной водой объем до 200 мл, нагревают до t = 80 0С на плитке.
Алюминиевый анод перед электролизом очищают наждачной бумагой, а затем обрабатывают 20 мл 20%-го раствора NaOH и промывают водой.Электроды погружают в раствор и проводят электролиз в течение 20–30 мин (при необходимости можно подогреть раствор с помощью спиртовки). Полноту осаждения меди определяют путем погружения в раствор оставшейся чистой поверхности платинового электрода.
После полного выделения меди, не размыкая цепи, электроды обмывают водой, затем разъединив – спиртом и эфиром. Катод с осадком высушивают над плиткой с закрытой спиралью, охлаждают и взвешивают. После взвешивания осадок с платинового электрода удаляют теплым раствором азотной кислоты. Электрод обмывают водой, спиртом, эфиром, высушивают и сдают лаборанту.
Рассчитывают содержание меди (в %) в дюралюминии.
2. ВОЛЬТАМПЕРОМЕТРИЯ
Вольтамперометрия основана на изучении поляризационных или вольтамперных кривых, которые получаются, если при электролизе раствора анализируемого вещества постепенно повышать напряжение и фиксировать при этом силу тока.
Классическая вольтамперометрия и получившая широкое распространение классическая полярография отличаются друг от друга тем, что в полярографии применяется ртутный электрод, а в вольтамперометрии – электроды других типов.
Полярография – это часть вольтамперометрии, в которой в качестве катода используется жидкий металлический ртутный электрод, поверхность которого периодически обновляется. В основе метода лежит явление концентрационной поляризации.
2.1. Прямые методы вольтамперометрии
2.1.1. Классическая полярография
Д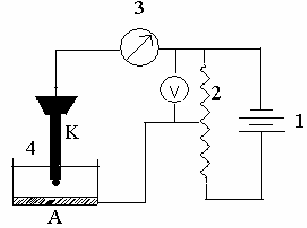
ля проведения полярографических исследований используется полярограф, принципиальная схема которого показана на рис. 5.
С помощью делителя напряжения 2 от внешнего источника постоянного тока 1 на электролизную ячейку 4 налагается медленно растущее напряжение со скоростью 0,1–0,2 В/мин. При этом в ячейке протекает ток, который фиксируется регистрирующим устройством 3 (гальванометр, микроамперметр, самописец).
Основными условиями полярографических исследований являются:
-
медленная скорость подачи напряжения;
-
п
Рис. 5. Принципиальная схема классического полярографа:
1 – источник постоянного тока;
2 – делитель напряжения;
3 – регистрирующее устройство;
4 – электролизная ячейка (К – катод (ртутная капля), А – анод)
оверхность катода должна быть во много раз меньше, чем поверхность анода (при катодной поляризации);
-
отсутствие перемешивания раствора для сохранения концентрационной поляризации ртутного капающего электрода (РКЭ).
Вольтамперная кривая
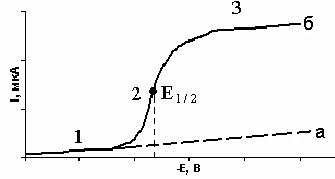
Полярограмма представляет собой графическое изображение зависимости силы тока от напряжения (потенциала), налагаемого на ячейку.
В
Рис. 6. Классическая полярограмма
отсутствие ионов, способных восстанавливаться на РКЭ, ток через раствор почти не протекает. Зависимость
I = f(E) в этом случае имеет вид прямой, с небольшим наклоном к оси абсцисс (а). При наличии в растворе ионов, способных к электровосстановлению, полярограмма имеет вид ступеньки (б) (рис. 6).
На полярограмме можно выделить 3 участка. В начале процесса (участок 1), при небольших значениях налагаемого напряжения, сила тока будет очень медленно возрастать.
Остаточный ток складывается из фарадеевского (If) и емкостного (конденсаторного) (Ic):
Iост = If +Ic.
Фарадеевский ток обусловлен разрядом электровосстанавливающихся примесей (плохо удаленного кислорода, либо следовых количеств тяжелых металлов). Емкостный ток обусловлен тем, что налагаемого потенциала недостаточно для разряда ионов деполяризатора. Ионы подходят к катоду, но не разряжаются, образуют у катода двойной электрический слой (ДЭС) – конденсатор. В случае стационарного ртутного катода после образования этого слоя ток немедленно прекратился бы. Однако ртуть постоянно вытекает из капилляра и для сообщения каждой новой капле заряда необходимо затратить новые порции электричества. При этом следует иметь в виду, что увеличение значения потенциала РКЭ связано с возрастанием плотности заряда на каждой последующей капле. Поэтому емкостный ток будет увеличиваться по мере возрастания потенциала катода.
Величина остаточного тока составляет порядка 1 мкА. При полярографировании малых концентраций анализируемых веществ (≤ 10-5 моль/л) величина остаточного тока становится сравнимой с величиной диффузионного тока. В этом случае необходимо учитывать остаточный ток.
При достижении потенциала восстановления ионов (участок 2) начинается их разряд на катоде:
Cd2+ + 2е + Hg Cd (Hg)
Сила тока резко возрастает, и будет определяться разностью концентраций деполяризатора в глубине раствора и приэлектродном слое:
I = ks(Cs– Cs0),
где ks – константа из уравнения Ильковича;
Cs –концентрация ионов в глубине раствора;
Cs0 –концентрация ионов в приэлектродном слое.
Образуется очень разбавленная амальгама кадмия, не препятствующая падению капли.
После достижения определенной величины, несмотря на увеличение налагаемого напряжения, сила тока остается постоянной, поскольку скорость восстановления ионов кадмия становится равной скорости диффузии. Этот ток называется предельным (участок 3).
Iдиф. = ksCs
Величина диффузионного тока прямо пропорциональна концентрации деполяризатора в массе раствора.
Предельный ток представляет собой сумму:
Iпред. = Iдиф. + Iмигр.
Диффузионный ток обусловлен диффузией ионов деполяризатора к поверхности РКЭ. Миграционный ток связан с электростатическими взаимодействиями ионов определяемого вещества (деполяризатора) с отрицательно заряженным РКЭ. Для устранения миграционного тока используют специальные фоновые электролиты, чаще всего растворимые соли щелочных, щелочно-земельных металлов и аммония. Так как концентрация ионов фона составляет порядка
0,1 М и превышает концентрацию деполяризатора в 103–104 раз, то именно они окружают РКЭ, экранируя действие его отрицательного заряда, но при этом не восстанавливаются, вследствие того, что для их восстановления необходимо более отрицательное значение потенциала. Таким образом, в присутствии фонового электролита перенос ионов деполяризатора осуществляется только за счет диффузии. В этом случае:
Iпред. = Iдиф.
Введение в раствор фонового электролита преследует следующие цели:
а) увеличение электропроводности раствора;
б) устранение миграционного тока;
в) определение нескольких ионов проводится путем варьирования состава фонового электролита, так как состав фонового электролита влияет на потенциал полуволны.
При наличии в растворе нескольких ионов металлов возможно их одновременное определение, если различия потенциалов полуволн составляют 0,2–0,3 В, при этом полярограмма представляет собой несколько волн – полярографический спектр (рис. 7).
П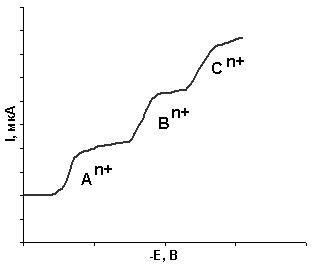
олярография – это метод одновременного качественного и количественного анализа. Основой качественного полярографического анализа является потенциал полуволны Е1/2, который зависит от природы иона, а также от состава фонового электролита.
М
Рис. 7. Полярографический спектр
смеси трех ионов
етодом классической полярографии можно определять концентрации вещества порядка 10-4–10-5 моль/л.
В основе количественного полярографического анализа лежит зависимость силы диффузионного тока от концентрации вещества в растворе. Эта зависимость описывается уравнением Ильковича:
Iдиф. = 605D1/2m2/31/ 6nC,
где D – коэффициент диффузии;
m – масса ртути, вытекающая в единицу времени, мг;
– период капания, 1/с;
n– количество электронов, участвующих в электродном процессе;
С – концентрация вещества, моль/л.
Если работают с одним и тем же капилляром и постоянной скоростью вытекания ртути, то произведение m2/31/6(константа капилляра) будет величиной постоянной, тогда уравнение Ильковича имеет следующий вид:
Iдиф. = KC
При расшифровке вольтамперной кривой в одинаковых условиях вместо силы тока можно использовать высоту волны. Для этого необходимо учесть все факторы, которые влияют на форму вольтамперной кривой и ее высоту. Такими факторами являются: емкостный ток, температура, растворенный кислород, максимумы на вольтамперных кривых. Значительное влияние оказывают два последних.
-
Кислород восстанавливается на РКЭ по двум ступеням:
первая ступень: от -0,1 В до -0,2 В
в кислой среде: О2 + 2Н+ + 2е Н2О2
в щелочной и нейтральной среде: О2 + 2Н2О 2Н2О2
вторая ступень: от -0,5 В до -1,3 В
в кислой среде: Н2О2 + 2Н+ + 2е 2Н2О
в щелочной и нейтральной среде: Н2О2 + 2е 2ОН-
Присутствие кислорода мешает определению веществ, особенно при их малой концентрации, когда его волна оказывается соизмеримой с волной определяемого вещества. Удаляют кислород, пропуская через раствор инертный газ (азот, аргон) в любых средах, в кислой среде можно использовать и углекислый газ СО2. В щелочных и нейтральных средах эффективно и быстро удаляет кислород Na2SO3. Можно применять и ментол.
-
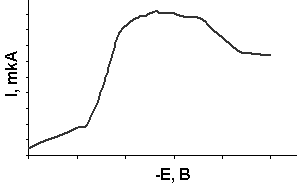
Максимумы на вольтамперных кривых. Очень часто вольтамперная кривая искажается максимумами, возникающими в той части кривой, которая соответствует началу предельного тока. Максимумы бывают разной формы – от пика до закругленного подъема. Возникновение максимумов на вольтамперных кривых мешает определению как потенциала полуволны, так и величины диффузионного тока.
Для объяснения причин возникновения максимумов необходимо рассмотреть зависимость величины поверхностного натяжения на границе раздела ртуть–раствор от налагаемого напряжения (электрокапиллярная кривая). Данная зависимость носит параболический характер с максимумом при -0,56 В (рис. 8).
П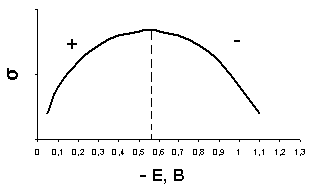
Рис. 8. Электрокапиллярная кривая
оверхность ртути в растворе ртутной соли, когда отсутствует потенциал извне, имеет положительный заряд. Отрицательно заряженные ионы или дипольные молекулы притягиваются к поверхности ртути и образуют двойной электрической слой (ДЭС) с определенным положительным потенциалом, которому соответствует невысокое поверхностное натяжение. При наложении отрицательного напряжения положительный заряд поверхности ртути уменьшается, что влечет за собой увеличение поверхностного натяжения до тех пор, пока ртуть не разрядится у максимума при -0,56 В. При этом потенциале исчезает заряд ДЭС и поверхностное натяжение достигает максимума. Потенциал максимума называется потенциалом электрокапиллярного нуля.
П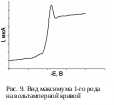 ри дальнейшем увеличении катодной поляризации ртуть заряжается отрицательно, и из раствора притягиваются уже положительно заряженные ионы и создается ДЭС, но противоположного знака. Поверхностное натяжение снова падает. Часть электрокапиллярной кривой до значения потенциала электрокапиллярного нуля обозначается знаком «+» (положительная ветвь), а после – знаком
ри дальнейшем увеличении катодной поляризации ртуть заряжается отрицательно, и из раствора притягиваются уже положительно заряженные ионы и создается ДЭС, но противоположного знака. Поверхностное натяжение снова падает. Часть электрокапиллярной кривой до значения потенциала электрокапиллярного нуля обозначается знаком «+» (положительная ветвь), а после – знаком
«–» (отрицательная ветвь).
Максимумы 1-го рода имеют характерный острый пик (рис. 9). Если потенциал полуволны деполяризатора, на волне которого имеется максимум, лежит на положительной ветви электрокапиллярной кривой, то он называется положительным. Если же максимум находится на отрицательной ветви – отрицательным.
В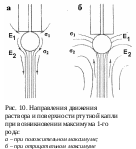 озникновение максимумов связано с тангенциальными движениями поверхности ртутной капли, которые вызывают перемешивание раствора и увеличивают подачу деполяризатора к электроду. Движение ртути возникает вследствие неравномерной поляризации ртутной капли (рис. 10).
озникновение максимумов связано с тангенциальными движениями поверхности ртутной капли, которые вызывают перемешивание раствора и увеличивают подачу деполяризатора к электроду. Движение ртути возникает вследствие неравномерной поляризации ртутной капли (рис. 10).
Плотность тока больше в нижней части капли, так как верхняя ее часть экранируется концом капилляра. Поэтому потенциал в разных точках неодинаков, что вызывает отличие в значениях поверхностного натяжения. Происходит движение ртути вдоль поверхности от участков с меньшим поверхностным натяжением к участкам с большим. Если максимум возникает в положительной части электрокапиллярной кривой, то движение поверхности ртутной капли и жидкости в электродном слое направлено сверху вниз:
Е1 Е2 и 1 2
В случае отрицательной части электрокапиллярной кривой (Е1 Е2,1 2) движение ртути и электролита направлено снизу вверх.
Максимумы 1-го рода устраняются введением в раствор поверхностно-активных веществ (ПАВ), которые адсорбируются на поверхности ртути. ПАВ подавляют максимумы в той области потенциалов, в которой они способны к адсорбции. Так, катионные ПАВ адсорбируются на отрицательно заряженной поверхности, анионные – на положительно заряженной поверхности, молекулярные ПАВ – в области электрокапиллярного нуля. ПАВ амфотерного характера (желатин, агар-агар) адсорбируются при всех потенциалах. Концентрация ПАВ подбирается экспериментально. Избыток ПАВ приводит к снижению диффузионного тока, поскольку препятствует диффузии деполяризатора к РКЭ.
Максимумы 2-го рода. Эти максимумы имеют пологий вид и захватывают широкую область значений потенциалов (рис. 11). Они имеют место при работе с быстро капающими капиллярами.
Максимумы 2-го рода возникают вследствие деформации капли при ее вытекании из капилляра. При росте капли струя ртути продолжается внутри капли до ее дна. Вследствие существования поверхностного натяжения эта струя не может выйти из самой капли, она отражается от поверхности и образует внутри капли завихрения, благодаря которым поверхность ртутной капли движется снизу вверх, увлекая за собой прилегающие слои электролита (рис. 12). Это явление наблюдается при всех потенциалах полярографической кривой и имеет максимальное значение в области электрокапиллярного нуля. Чтобы устранить максимумы 2-го рода, недостаточно добавлять к анализируемому раствору ПАВ, необходимо подбирать высоту ртутного столба, регулировать период капания ртути, менять капилляр.
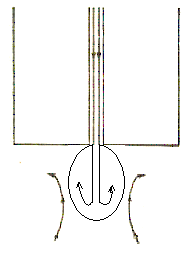
Ри с. 11. Вид максимума 2-го рода
на вольтамперной кривой
Рис. 12. Направления движения капли и раствора при возникновении максимумов 2-го рода
2.1.2. Осциллографическая полярография
В осциллографической полярографии скорость подачи напряжения составляет десятки вольт в секунду. При таком быстром изменении напряжения и силы тока для регистрации кривых удобнее пользоваться безынерционным прибором – осциллографом.
Основная часть осциллографа – это электронно-лучевая трубка. Катод в этой трубке, подогреваемый специальной спиралью, эмитирует пучок электронов, которые разгоняются в электрическом поле между катодом и анодом. Этот пучок проходит через две пары взаимно перпендикулярных пластин и падает на люминесцирующий экран. Причем горизонтальные пластины отклоняют луч в вертикальном направлении, а вертикальные пластины – в горизонтальном.
На горизонтальные пластины осциллографа налагается напряжение, пропорциональное силе тока в цепи, а на вертикальные – напряжение между электродами в ячейке.
Схема осциллографической установки приведена на рис. 13.
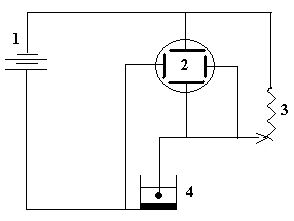
Рис. 13. Принципиальная схема осциллографического полярографа.
1– источник постоянного тока;
2 – осциллограф;
3 – делитель напряжения;
4 – электролизная ячейка
Н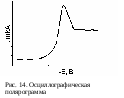 а экране осциллографа наблюдают кривую зависимости силы тока от подаваемого напряжения (рис. 14). Максимальная высота пика прямо пропорциональна концентрации деполяризатора, а потенциал максимума характеризует природу определяемого вещества.
а экране осциллографа наблюдают кривую зависимости силы тока от подаваемого напряжения (рис. 14). Максимальная высота пика прямо пропорциональна концентрации деполяризатора, а потенциал максимума характеризует природу определяемого вещества.
Уравнение максимального тока осциллографической кривой для линейной диффузии было выведено Рэндлсом и Шевчиком:
Imax = kn3/2D1/2AV1/2C, где
А – поверхность электрода, мм2, А = 0,85(m)2/3;
V – скорость подачи напряжения, В/с;
k – сложная константа, зависящая от характера электрохимического процесса (так, при 25 ºС k = 2,69·105).
Факторами, снижающими чувствительность осциллографии, являются емкостные токи. Оказалось, что увеличению тока пика с возрастанием скорости подачи напряжения соответствует еще большее увеличение емкостного тока. Ток пика растет пропорционально корню квадратному из скорости подачи напряжения. Емкостный ток увеличивается пропорционально скорости подачи напряжения:
Iемк. = ACD(U)V, а Imax=f (V1/2),
где А – поверхность электрода, мм2;
CD(U) – удельная диффузионная емкость, мкФ;
V – скорость подачи напряжения, В/с.
При больших скоростях подачи напряжения кривая настолько искажена, что использовать ее невозможно.
Методом осциллографической полярографии определяют концентрации веществ порядка 10-5–10-7 моль/л.
Количественный полярографический анализ
Уравнение Ильковича для нахождения концентрации деполяризатора обычно не применяют, так как для этого необходимо знать величину коэффициента диффузии, зависящую от условий регистрации полярограммы (от состава и концентрации фона, температуры и др.). В практической работе обычно используют для нахождения концентрации способ стандартов, градуировочного графика или способ добавок.
А. Определение концентрации деполяризатора по способу стандартов
Для определения концентрации деполяризатора сначала записывают полярограмму раствора с точно известной концентрацией и определяют высоту волны. Затем производят запись полярограммы анализируемого раствора и также определяют высоту волны. Зная концентрацию и высоту волны для стандартного раствора, и высоту волны для анализируемого, находят концентрацию деполяризатора в анализируемом растворе:
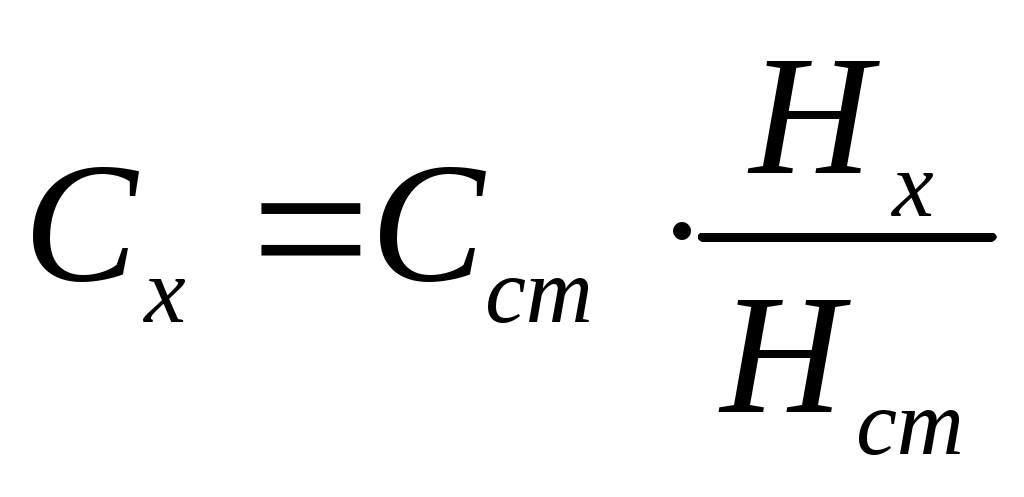
При регистрации полярограмм необходимо соблюдать следующие условия: все измерения должны быть получены с одним капилляром, при одной постоянной высоте столба ртути над капилляром и при одинаковой чувствительности прибора, состав стандартного и исследуемого растворов должны быть идентичными. Такой способ определения концентрации достаточно экспрессный, но его нельзя использовать при анализе сложных объектов без предварительного выделения определяемого элемента.
Б. Определение концентрации деполяризатора по способу градуировочного графика
Для построения градуировочного графика регистрируют полярограммы стандартных растворов определяемого деполяризатора (4–5 растворов в изучаемом диапазоне концентраций) и по полученным экспериментальным данным строят зависимость высоты волны от концентрации.
При регистрации полярограмм стандартных и анализируемых растворов необходимо соблюдать те же условия, что и при использовании способа стандартов.
В. Определение концентрации деполяризатора по способу добавок
Для анализа сложных по составу объектов (сталей, сплавов, почв и т. п.) целесообразно пользоваться способом добавок. Он применим в диапазоне линейной зависимости высоты волны от концентрации деполяризатора.
В ячейку помещают определенное количество исследуемого раствора, регистрируют полярограмму, затем непосредственно в ячейку добавляют стандартный раствор определяемого вещества с таким расчетом, чтобы высота волны увеличилась вдвое. Объем добавки не должен превышать 0,5 мл. После продувки инертным газом регистрируют полярограмму в тех же условиях. Измеряют высоты волн и строят графическую зависимость высоты волны Н от концентрации добавки (рис. 15). Экстраполируя прямую, находят концентрацию исследуемого раствора в тех же единицах, что и концентрация добавки.
Общие указания по работе на полярографической установке
1. Современный полярограф – сложный прибор, поэтому перед проведением эксперимента необходимо:
• внимательно ознакомиться с описанием прибора и инструкцией по его использованию; особое внимание обратить на управляющие ручки, отмеченные в тексте;
• понять цель работы и порядок ее выполнения;
• обсудить с преподавателем порядок выполнения работы и функции управляющих органов прибора;
• c разрешения преподавателя включить прибор и подготовить необходимые растворы.
2. Электронные полярографические приборы питаются от сети переменного тока 220 В, для питания отдельных блоков приборов используют и более высокое напряжение. Поэтому приборы должны быть хорошо заземлены.
В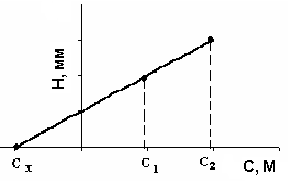
случае малейших неполадок следует немедленно обращаться к преподавателю или лаборанту.
В
Рис. 15. Зависимость высоты полярографической волны от концентрации добавки
полярографии используют ртутные капающие электроды и электроды сравнения с большой поверхностью ртути; в процессе работы ртуть накапливается на дне электролизера. Необходимо помнить, что пары ртути опасны для организма. Однако при тщательном выполнении инструкции и аккуратной работе возможность заражения воздуха лаборатории сводится к минимуму.
Запрещается
• использовать нагревательные приборы вблизи ртутных ячеек;
• оставлять открытой поверхность ртути в электролизере (всегда должен быть слой жидкости – воды или раствора);
• оставлять ртутный капающий электрод на воздухе, не подставив под капилляр стакан с водой или электролизер с раствором;
• работать с ртутью вне специального поддона;
• сливать отработанные растворы в раковину. Все растворы из электролизеров следует сливать только в специальные банки для слива, находящиеся в том же поддоне. Электроды промывают дистиллированной водой из промывалки, подставив под электроды стакан. При обнаружении капель ртути в поддоне или при проливании ртути надо немедленно сообщить лаборанту и преподавателю.
ЛАБОРАТОРНАЯ РАБОТА 3
Определение ионов таллия и цинка при совместном
присутствии методом классической полярографии
Цель работы: 1. Изучить влияние мешающих факторов (растворенный кислород и максимум 1-го рода) на вольтамперную кривую.
2. Провести качественный и количественный полярографический анализ ионов металлов при их совместном присутствии.
А. Регистрация полярограммы кислорода. Удаление растворенного кислорода и максимума 1-го рода
Проведению полярографических измерений мешают растворенный кислород и максимумы 1-го рода. В разбавленных водных растворах солей растворяется приблизительно 2·l0-4 M кислорода. Кислород восстанавливается на ртутном электроде. На первой волне, помимо восстановления кислорода, наблюдается полярографический максимум 1-го рода, особенно ярко выраженный в разбавленных фоновых растворах (рис. 16, кривая 1). Полярографические максимумы мешают обработке полярограмм, поэтому их устраняют введением ПАВ. На полярограмме по-прежнему остаются волны восстановления кислорода (рис. 16, кривая 2).
П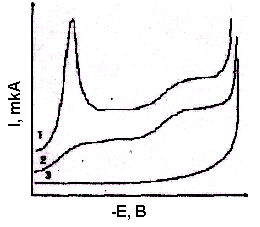
еред регистрацией полярограммы удаляют растворенный кислород. Из щелочных и нейтральных растворов кислород довольно быстро удаляется восстановлением сульфит-ионами (О2 + 2Na2SО3 → 2Na2SО4).
Д
Рис. 16. Полярограммы кислорода на фоне 0,01 М КС1
1 – до прибавления желатина;
2 – после добавления желатина;
3 – полярограмма 0,01 М КС1 после
удаления кислорода с помощью Na2SО3
обавление 0,1 г Na2SО3 в 100 мл раствора достаточно для удаления кислорода в течение 5 мин (рис. 16, кривая 3).
Реагенты и аппаратура
Хлорид калия, 0,01 М раствор.
Желатин, 0,1%-ный раствор.
Сульфит натрия, кристаллический.
Ячейка с ртутным капающим электродом.
1. Полярограф LP-60 готовят к работе согласно описанию прибора. Для проверки работоспособности прибора в измерительную цепь вместо ячейки подключают сопротивление (100 кОм) и, включив развертку постоянного напряжения, регистрируют изменение тока в цепи в интервале потенциалов от 0 до -1,2 В (скорость развертки 200 мВ/мин, чувствительность 1/200).
Если прибор исправен, самописец регистрирует прямую линию с наклоном (угол 450) при диапазоне тока (чувствительности) 1/200.
2. Выполнение работы. Электролизер тщательно промывают дистиллированной водой (все сливы собирают в специальную склянку!) и помещают в него 0,01М KCI. Ртутный капающий электрод опускает в ячейку и вынимает из ячейки только преподаватель или лаборант! Кончик капилляра должен быть обязательно погружен в раствор.
Записывают классическую полярограмму (скорость развертки потенциала 200 мВ/мин, чувствительность 1/100) от 0 до -1,5 В.
Ознакомившись с полярограммой (она должна быть похожа на приведенную на рис. 16 кривую 1), убеждаются, что максимум 1-го рода можно устранить с помощью желатина. Для этого добавляют в электролизер 2 капли раствора желатина и записывают полярограмму. Убедившись, что максимум исчез, и на полярограмме присутствуют две волны восстановления кислорода (рис. 16, кривая 2), приступают к удалению растворенного кислорода.
К раствору в электролизере прибавляют примерно 0,1 г Na2SО3 (половина шпателя), перемешивают и через 5 мин записывают полярограмму. Сравнивают полярограмму (она должна быть похожа на приведенную на рис. 16 кривую 3) с предыдущими и убеждаются, что кислород из раствора удален.
Б. Регистрация и расшифровка полярограммы смеси деполяризаторов (полярографический спектр)
Реагенты и аппаратура
Стандартные 0,01 М растворы Т1(1), Zn(II).
0,1 М раствор KCl.
Сульфит натрия, кристаллический.
Желатин, 0,1%-ный раствор.
Полярограф LP-60.
Ячейка с ртутным капающим
электродом.
-
Выполнение работы. К полученному у преподавателя раствору, содержащему ионы Т1(I) и Zn(II), прибавляют примерно 0,1 г сульфита натрия, 2 капли раствора желатины и разбавляют до метки фоновым электролитом (0,1 М KCl).
Электролизер тщательно промывают сначала водой, затем анализируемым раствором, чтобы избежать разбавления. Помещают в электролизер исследуемый раствор так, чтобы кончик капилляра был погружен в раствор.
Записывают классическую полярограмму при непрерывном режиме регистрации тока от -0,2 В до -1,3 В при скорости развертки потенциала 200 мВ/мин (чувствительность 1/100).
Для расшифровки полярограмм очень важно отметить начало записи полярограммы и величину начального потенциала.
2. Расшифровка полярограмм и представление результатов:
а) измеряют высоты всех волн (мм) на полярограмме, проведя касательные ко всем трем участкам волны и опустив вертикаль из верхней точки пересечения на нижнюю касательную;
б) по способу стандартов рассчитывают концентрации ионов таллия и цинка в анализируемом растворе;
в) измеряют величины Е1/2 всех волн с учетом величины начального потенциала и масштаба диаграммы по оси потенциалов.
Найденные значения Е1/2 сравнивают с табличными для 0,1 М раствора KCl и определяют, какие деполяризаторы находятся в растворе.
ЛАБОРАТОРНАЯ РАБОТА 4
Определение малых концентраций ионов кадмия
на осциллографическом полярографе
1 2 3 4
Проба бронзы.
HNO3 (1:1).
15%-ный растворNH4NO3.
H2SO4 (1:1).
10%-ный раствор NH4OH.
Спирт этиловый.
Диэтиловый эфир.
HNO3 концентрированная.
Платиновые электроды – 2 шт.
Аккумулятор на 4 В.
Методика выполнения работы: навеску 0,1 г бронзы помещают в стакан и растворяют в азотной кислоте (1:1). Кислоту приливают постепенно, порциями по 2–3 мл. Растворение ведут при нагревании (под тягой) на плитке. К полученному раствору добавляют 100 мл горячей дистиллированной воды, 20 мл 15%-го раствора нитрата аммония и кипятят 10–15 мин. Затем раствор упаривают до объема 100–120 мл и добавляют к нему 2,5 мл азотной кислоты 1:1.
В полученный раствор опускают электроды, предварительно взвешенные на аналитических весах. Электролиз ведут при напряжении 2,2–2,4 В и силе тока 1,8–3,0 А. Схема установки электролиза представлена на рис. 4.
Ч
ерез 30–35 мин добавляют 2 мл серной кислоты (1:1) и, не прекращая электролиза, частично нейтрализуют раствор 2,5–3,0 мл 10%-го раствора NH4OH. Продолжают электролиз еще в течение 20–30 мин. Затем приливают в электролизер такое количество воды, чтобы уровень жидкости повысился на 1–1,5 см и проверяют выделение меди на свежей поверхности электрода. Е Рис. 4. Схема установки
для электролиза:
1 – аккумулятор на 4 В;
2 – амперметр;
3 – вольтметр;
4 – реостат;
5 – установка для электролиза с платиновыми электродами.
сли медь не выделяется из раствора, то, не прерывая тока, убирают стакан с раствором и промывают электроды, подставляя стакан с дистиллированной водой. После этого размыкают цепь, снимают электроды и промывают их спиртом, эфиром, высушивают над электроплиткой и взвешивают.
По окончании работы установку разбирают, электроды промывают: катод в концентрированной азотной кислоте, анод – в концентрированной азотной кислоте с добавлением 0,1 н щавелевой кислоты, затем в воде, спирте, эфире. После промывания электроды высушивают, взвешивают и сдают лаборанту.
Рассчитывают процентное содержание меди и свинца в бронзе.
ЛАБОРАТОРНАЯ РАБОТА 2
Определение меди в дюралюминии методом внутреннего электролиза в дюралюминии
Цель работы: определить процентное содержание меди в сплаве дюралюминия.
Реагенты и аппаратура
Проба дюралюминия.
20%-ный раствор NaOH.
2н раствор H2SO4.
HNO3 концентрированная.
Спирт этиловый.
Диэтиловый эфир.
Платиновый электрод – 1 шт.
Алюминиевый электрод – 1 шт.
Методика выполнения работы: в небольшом химическом стакане растворяют 0,1 г сплава дюралюминия в возможно малом количестве 20%-го едкого натра при нагревании (на плитке под тягой). Алюминий переходит в раствор, а медь, железо, марганец остаются в осадке:
2Al + 2NaOH + 6H2O → 2Na[Al(OH)4] + 3H2↑
Осадок растворяют при охлаждении в концентрированной азотной кислоте, которую добавляют по каплям (не более 1,5 мл):
Na[Al(OH)4] + 4 HNO3 → Al(NO3)3 + NaNO3 + 4H2O
Cu + 4HNO3 → Cu(NO3)2 + 2NO2↑ + 2H2O
После нагревания осадка и растворения его, к исследуемому раствору добавляют 100 мл 2 н серной кислоты, доводят дистиллированной водой объем до 200 мл, нагревают до t = 80 0С на плитке.
Алюминиевый анод перед электролизом очищают наждачной бумагой, а затем обрабатывают 20 мл 20%-го раствора NaOH и промывают водой.Электроды погружают в раствор и проводят электролиз в течение 20–30 мин (при необходимости можно подогреть раствор с помощью спиртовки). Полноту осаждения меди определяют путем погружения в раствор оставшейся чистой поверхности платинового электрода.
После полного выделения меди, не размыкая цепи, электроды обмывают водой, затем разъединив – спиртом и эфиром. Катод с осадком высушивают над плиткой с закрытой спиралью, охлаждают и взвешивают. После взвешивания осадок с платинового электрода удаляют теплым раствором азотной кислоты. Электрод обмывают водой, спиртом, эфиром, высушивают и сдают лаборанту.
Рассчитывают содержание меди (в %) в дюралюминии.
2. ВОЛЬТАМПЕРОМЕТРИЯ
Вольтамперометрия основана на изучении поляризационных или вольтамперных кривых, которые получаются, если при электролизе раствора анализируемого вещества постепенно повышать напряжение и фиксировать при этом силу тока.
Классическая вольтамперометрия и получившая широкое распространение классическая полярография отличаются друг от друга тем, что в полярографии применяется ртутный электрод, а в вольтамперометрии – электроды других типов.
Полярография – это часть вольтамперометрии, в которой в качестве катода используется жидкий металлический ртутный электрод, поверхность которого периодически обновляется. В основе метода лежит явление концентрационной поляризации.
2.1. Прямые методы вольтамперометрии
2.1.1. Классическая полярография
Д
ля проведения полярографических исследований используется полярограф, принципиальная схема которого показана на рис. 5.
С помощью делителя напряжения 2 от внешнего источника постоянного тока 1 на электролизную ячейку 4 налагается медленно растущее напряжение со скоростью 0,1–0,2 В/мин. При этом в ячейке протекает ток, который фиксируется регистрирующим устройством 3 (гальванометр, микроамперметр, самописец).
Основными условиями полярографических исследований являются:
-
медленная скорость подачи напряжения; -
п
Рис. 5. Принципиальная схема классического полярографа:
1 – источник постоянного тока;
2 – делитель напряжения;
3 – регистрирующее устройство;
4 – электролизная ячейка (К – катод (ртутная капля), А – анод)
оверхность катода должна быть во много раз меньше, чем поверхность анода (при катодной поляризации); -
отсутствие перемешивания раствора для сохранения концентрационной поляризации ртутного капающего электрода (РКЭ).
Вольтамперная кривая
Полярограмма представляет собой графическое изображение зависимости силы тока от напряжения (потенциала), налагаемого на ячейку.
В
Рис. 6. Классическая полярограмма
отсутствие ионов, способных восстанавливаться на РКЭ, ток через раствор почти не протекает. Зависимость
I = f(E) в этом случае имеет вид прямой, с небольшим наклоном к оси абсцисс (а). При наличии в растворе ионов, способных к электровосстановлению, полярограмма имеет вид ступеньки (б) (рис. 6).
На полярограмме можно выделить 3 участка. В начале процесса (участок 1), при небольших значениях налагаемого напряжения, сила тока будет очень медленно возрастать.
Остаточный ток складывается из фарадеевского (If) и емкостного (конденсаторного) (Ic):
Iост = If +Ic.
Фарадеевский ток обусловлен разрядом электровосстанавливающихся примесей (плохо удаленного кислорода, либо следовых количеств тяжелых металлов). Емкостный ток обусловлен тем, что налагаемого потенциала недостаточно для разряда ионов деполяризатора. Ионы подходят к катоду, но не разряжаются, образуют у катода двойной электрический слой (ДЭС) – конденсатор. В случае стационарного ртутного катода после образования этого слоя ток немедленно прекратился бы. Однако ртуть постоянно вытекает из капилляра и для сообщения каждой новой капле заряда необходимо затратить новые порции электричества. При этом следует иметь в виду, что увеличение значения потенциала РКЭ связано с возрастанием плотности заряда на каждой последующей капле. Поэтому емкостный ток будет увеличиваться по мере возрастания потенциала катода.
Величина остаточного тока составляет порядка 1 мкА. При полярографировании малых концентраций анализируемых веществ (≤ 10-5 моль/л) величина остаточного тока становится сравнимой с величиной диффузионного тока. В этом случае необходимо учитывать остаточный ток.
При достижении потенциала восстановления ионов (участок 2) начинается их разряд на катоде:
Cd2+ + 2е + Hg Cd (Hg)
Сила тока резко возрастает, и будет определяться разностью концентраций деполяризатора в глубине раствора и приэлектродном слое:
I = ks(Cs– Cs0),
где ks – константа из уравнения Ильковича;
Cs –концентрация ионов в глубине раствора;
Cs0 –концентрация ионов в приэлектродном слое.
Образуется очень разбавленная амальгама кадмия, не препятствующая падению капли.
После достижения определенной величины, несмотря на увеличение налагаемого напряжения, сила тока остается постоянной, поскольку скорость восстановления ионов кадмия становится равной скорости диффузии. Этот ток называется предельным (участок 3).
Iдиф. = ksCs
Величина диффузионного тока прямо пропорциональна концентрации деполяризатора в массе раствора.
Предельный ток представляет собой сумму:
Iпред. = Iдиф. + Iмигр.
Диффузионный ток обусловлен диффузией ионов деполяризатора к поверхности РКЭ. Миграционный ток связан с электростатическими взаимодействиями ионов определяемого вещества (деполяризатора) с отрицательно заряженным РКЭ. Для устранения миграционного тока используют специальные фоновые электролиты, чаще всего растворимые соли щелочных, щелочно-земельных металлов и аммония. Так как концентрация ионов фона составляет порядка
0,1 М и превышает концентрацию деполяризатора в 103–104 раз, то именно они окружают РКЭ, экранируя действие его отрицательного заряда, но при этом не восстанавливаются, вследствие того, что для их восстановления необходимо более отрицательное значение потенциала. Таким образом, в присутствии фонового электролита перенос ионов деполяризатора осуществляется только за счет диффузии. В этом случае:
Iпред. = Iдиф.
Введение в раствор фонового электролита преследует следующие цели:
а) увеличение электропроводности раствора;
б) устранение миграционного тока;
в) определение нескольких ионов проводится путем варьирования состава фонового электролита, так как состав фонового электролита влияет на потенциал полуволны.
При наличии в растворе нескольких ионов металлов возможно их одновременное определение, если различия потенциалов полуволн составляют 0,2–0,3 В, при этом полярограмма представляет собой несколько волн – полярографический спектр (рис. 7).
П
олярография – это метод одновременного качественного и количественного анализа. Основой качественного полярографического анализа является потенциал полуволны Е1/2, который зависит от природы иона, а также от состава фонового электролита.
М
Рис. 7. Полярографический спектр
смеси трех ионов
етодом классической полярографии можно определять концентрации вещества порядка 10-4–10-5 моль/л.
В основе количественного полярографического анализа лежит зависимость силы диффузионного тока от концентрации вещества в растворе. Эта зависимость описывается уравнением Ильковича:
Iдиф. = 605D1/2m2/31/ 6nC,
где D – коэффициент диффузии;
m – масса ртути, вытекающая в единицу времени, мг;
– период капания, 1/с;
n– количество электронов, участвующих в электродном процессе;
С – концентрация вещества, моль/л.
Если работают с одним и тем же капилляром и постоянной скоростью вытекания ртути, то произведение m2/31/6(константа капилляра) будет величиной постоянной, тогда уравнение Ильковича имеет следующий вид:
Iдиф. = KC
При расшифровке вольтамперной кривой в одинаковых условиях вместо силы тока можно использовать высоту волны. Для этого необходимо учесть все факторы, которые влияют на форму вольтамперной кривой и ее высоту. Такими факторами являются: емкостный ток, температура, растворенный кислород, максимумы на вольтамперных кривых. Значительное влияние оказывают два последних.
-
Кислород восстанавливается на РКЭ по двум ступеням:
первая ступень: от -0,1 В до -0,2 В
в кислой среде: О2 + 2Н+ + 2е Н2О2
в щелочной и нейтральной среде: О2 + 2Н2О 2Н2О2
вторая ступень: от -0,5 В до -1,3 В
в кислой среде: Н2О2 + 2Н+ + 2е 2Н2О
в щелочной и нейтральной среде: Н2О2 + 2е 2ОН-
Присутствие кислорода мешает определению веществ, особенно при их малой концентрации, когда его волна оказывается соизмеримой с волной определяемого вещества. Удаляют кислород, пропуская через раствор инертный газ (азот, аргон) в любых средах, в кислой среде можно использовать и углекислый газ СО2. В щелочных и нейтральных средах эффективно и быстро удаляет кислород Na2SO3. Можно применять и ментол.
-
Максимумы на вольтамперных кривых. Очень часто вольтамперная кривая искажается максимумами, возникающими в той части кривой, которая соответствует началу предельного тока. Максимумы бывают разной формы – от пика до закругленного подъема. Возникновение максимумов на вольтамперных кривых мешает определению как потенциала полуволны, так и величины диффузионного тока.
Для объяснения причин возникновения максимумов необходимо рассмотреть зависимость величины поверхностного натяжения на границе раздела ртуть–раствор от налагаемого напряжения (электрокапиллярная кривая). Данная зависимость носит параболический характер с максимумом при -0,56 В (рис. 8).
П
Рис. 8. Электрокапиллярная кривая
оверхность ртути в растворе ртутной соли, когда отсутствует потенциал извне, имеет положительный заряд. Отрицательно заряженные ионы или дипольные молекулы притягиваются к поверхности ртути и образуют двойной электрической слой (ДЭС) с определенным положительным потенциалом, которому соответствует невысокое поверхностное натяжение. При наложении отрицательного напряжения положительный заряд поверхности ртути уменьшается, что влечет за собой увеличение поверхностного натяжения до тех пор, пока ртуть не разрядится у максимума при -0,56 В. При этом потенциале исчезает заряд ДЭС и поверхностное натяжение достигает максимума. Потенциал максимума называется потенциалом электрокапиллярного нуля.
П
ри дальнейшем увеличении катодной поляризации ртуть заряжается отрицательно, и из раствора притягиваются уже положительно заряженные ионы и создается ДЭС, но противоположного знака. Поверхностное натяжение снова падает. Часть электрокапиллярной кривой до значения потенциала электрокапиллярного нуля обозначается знаком «+» (положительная ветвь), а после – знаком «–» (отрицательная ветвь).
Максимумы 1-го рода имеют характерный острый пик (рис. 9). Если потенциал полуволны деполяризатора, на волне которого имеется максимум, лежит на положительной ветви электрокапиллярной кривой, то он называется положительным. Если же максимум находится на отрицательной ветви – отрицательным.
В
озникновение максимумов связано с тангенциальными движениями поверхности ртутной капли, которые вызывают перемешивание раствора и увеличивают подачу деполяризатора к электроду. Движение ртути возникает вследствие неравномерной поляризации ртутной капли (рис. 10). Плотность тока больше в нижней части капли, так как верхняя ее часть экранируется концом капилляра. Поэтому потенциал в разных точках неодинаков, что вызывает отличие в значениях поверхностного натяжения. Происходит движение ртути вдоль поверхности от участков с меньшим поверхностным натяжением к участкам с большим. Если максимум возникает в положительной части электрокапиллярной кривой, то движение поверхности ртутной капли и жидкости в электродном слое направлено сверху вниз:
Е1 Е2 и 1 2
В случае отрицательной части электрокапиллярной кривой (Е1 Е2,1 2) движение ртути и электролита направлено снизу вверх.
Максимумы 1-го рода устраняются введением в раствор поверхностно-активных веществ (ПАВ), которые адсорбируются на поверхности ртути. ПАВ подавляют максимумы в той области потенциалов, в которой они способны к адсорбции. Так, катионные ПАВ адсорбируются на отрицательно заряженной поверхности, анионные – на положительно заряженной поверхности, молекулярные ПАВ – в области электрокапиллярного нуля. ПАВ амфотерного характера (желатин, агар-агар) адсорбируются при всех потенциалах. Концентрация ПАВ подбирается экспериментально. Избыток ПАВ приводит к снижению диффузионного тока, поскольку препятствует диффузии деполяризатора к РКЭ.
Максимумы 2-го рода. Эти максимумы имеют пологий вид и захватывают широкую область значений потенциалов (рис. 11). Они имеют место при работе с быстро капающими капиллярами.
Максимумы 2-го рода возникают вследствие деформации капли при ее вытекании из капилляра. При росте капли струя ртути продолжается внутри капли до ее дна. Вследствие существования поверхностного натяжения эта струя не может выйти из самой капли, она отражается от поверхности и образует внутри капли завихрения, благодаря которым поверхность ртутной капли движется снизу вверх, увлекая за собой прилегающие слои электролита (рис. 12). Это явление наблюдается при всех потенциалах полярографической кривой и имеет максимальное значение в области электрокапиллярного нуля. Чтобы устранить максимумы 2-го рода, недостаточно добавлять к анализируемому раствору ПАВ, необходимо подбирать высоту ртутного столба, регулировать период капания ртути, менять капилляр.
| Ри с. 11. Вид максимума 2-го рода на вольтамперной кривой | Рис. 12. Направления движения капли и раствора при возникновении максимумов 2-го рода |
2.1.2. Осциллографическая полярография
В осциллографической полярографии скорость подачи напряжения составляет десятки вольт в секунду. При таком быстром изменении напряжения и силы тока для регистрации кривых удобнее пользоваться безынерционным прибором – осциллографом.
Основная часть осциллографа – это электронно-лучевая трубка. Катод в этой трубке, подогреваемый специальной спиралью, эмитирует пучок электронов, которые разгоняются в электрическом поле между катодом и анодом. Этот пучок проходит через две пары взаимно перпендикулярных пластин и падает на люминесцирующий экран. Причем горизонтальные пластины отклоняют луч в вертикальном направлении, а вертикальные пластины – в горизонтальном.
На горизонтальные пластины осциллографа налагается напряжение, пропорциональное силе тока в цепи, а на вертикальные – напряжение между электродами в ячейке.
Схема осциллографической установки приведена на рис. 13.
Рис. 13. Принципиальная схема осциллографического полярографа.
1– источник постоянного тока;
2 – осциллограф;
3 – делитель напряжения;
4 – электролизная ячейка
Н
а экране осциллографа наблюдают кривую зависимости силы тока от подаваемого напряжения (рис. 14). Максимальная высота пика прямо пропорциональна концентрации деполяризатора, а потенциал максимума характеризует природу определяемого вещества.Уравнение максимального тока осциллографической кривой для линейной диффузии было выведено Рэндлсом и Шевчиком:
Imax = kn3/2D1/2AV1/2C, где
А – поверхность электрода, мм2, А = 0,85(m)2/3;
V – скорость подачи напряжения, В/с;
k – сложная константа, зависящая от характера электрохимического процесса (так, при 25 ºС k = 2,69·105).
Факторами, снижающими чувствительность осциллографии, являются емкостные токи. Оказалось, что увеличению тока пика с возрастанием скорости подачи напряжения соответствует еще большее увеличение емкостного тока. Ток пика растет пропорционально корню квадратному из скорости подачи напряжения. Емкостный ток увеличивается пропорционально скорости подачи напряжения:
Iемк. = ACD(U)V, а Imax=f (V1/2),
где А – поверхность электрода, мм2;
CD(U) – удельная диффузионная емкость, мкФ;
V – скорость подачи напряжения, В/с.
При больших скоростях подачи напряжения кривая настолько искажена, что использовать ее невозможно.
Методом осциллографической полярографии определяют концентрации веществ порядка 10-5–10-7 моль/л.
Количественный полярографический анализ
Уравнение Ильковича для нахождения концентрации деполяризатора обычно не применяют, так как для этого необходимо знать величину коэффициента диффузии, зависящую от условий регистрации полярограммы (от состава и концентрации фона, температуры и др.). В практической работе обычно используют для нахождения концентрации способ стандартов, градуировочного графика или способ добавок.
А. Определение концентрации деполяризатора по способу стандартов
Для определения концентрации деполяризатора сначала записывают полярограмму раствора с точно известной концентрацией и определяют высоту волны. Затем производят запись полярограммы анализируемого раствора и также определяют высоту волны. Зная концентрацию и высоту волны для стандартного раствора, и высоту волны для анализируемого, находят концентрацию деполяризатора в анализируемом растворе:
При регистрации полярограмм необходимо соблюдать следующие условия: все измерения должны быть получены с одним капилляром, при одной постоянной высоте столба ртути над капилляром и при одинаковой чувствительности прибора, состав стандартного и исследуемого растворов должны быть идентичными. Такой способ определения концентрации достаточно экспрессный, но его нельзя использовать при анализе сложных объектов без предварительного выделения определяемого элемента.
Б. Определение концентрации деполяризатора по способу градуировочного графика
Для построения градуировочного графика регистрируют полярограммы стандартных растворов определяемого деполяризатора (4–5 растворов в изучаемом диапазоне концентраций) и по полученным экспериментальным данным строят зависимость высоты волны от концентрации.
При регистрации полярограмм стандартных и анализируемых растворов необходимо соблюдать те же условия, что и при использовании способа стандартов.
В. Определение концентрации деполяризатора по способу добавок
Для анализа сложных по составу объектов (сталей, сплавов, почв и т. п.) целесообразно пользоваться способом добавок. Он применим в диапазоне линейной зависимости высоты волны от концентрации деполяризатора.
В ячейку помещают определенное количество исследуемого раствора, регистрируют полярограмму, затем непосредственно в ячейку добавляют стандартный раствор определяемого вещества с таким расчетом, чтобы высота волны увеличилась вдвое. Объем добавки не должен превышать 0,5 мл. После продувки инертным газом регистрируют полярограмму в тех же условиях. Измеряют высоты волн и строят графическую зависимость высоты волны Н от концентрации добавки (рис. 15). Экстраполируя прямую, находят концентрацию исследуемого раствора в тех же единицах, что и концентрация добавки.
Общие указания по работе на полярографической установке
1. Современный полярограф – сложный прибор, поэтому перед проведением эксперимента необходимо:
• внимательно ознакомиться с описанием прибора и инструкцией по его использованию; особое внимание обратить на управляющие ручки, отмеченные в тексте;
• понять цель работы и порядок ее выполнения;
• обсудить с преподавателем порядок выполнения работы и функции управляющих органов прибора;
• c разрешения преподавателя включить прибор и подготовить необходимые растворы.
2. Электронные полярографические приборы питаются от сети переменного тока 220 В, для питания отдельных блоков приборов используют и более высокое напряжение. Поэтому приборы должны быть хорошо заземлены.
В
случае малейших неполадок следует немедленно обращаться к преподавателю или лаборанту.
В
Рис. 15. Зависимость высоты полярографической волны от концентрации добавки
полярографии используют ртутные капающие электроды и электроды сравнения с большой поверхностью ртути; в процессе работы ртуть накапливается на дне электролизера. Необходимо помнить, что пары ртути опасны для организма. Однако при тщательном выполнении инструкции и аккуратной работе возможность заражения воздуха лаборатории сводится к минимуму.
Запрещается
• использовать нагревательные приборы вблизи ртутных ячеек;
• оставлять открытой поверхность ртути в электролизере (всегда должен быть слой жидкости – воды или раствора);
• оставлять ртутный капающий электрод на воздухе, не подставив под капилляр стакан с водой или электролизер с раствором;
• работать с ртутью вне специального поддона;
• сливать отработанные растворы в раковину. Все растворы из электролизеров следует сливать только в специальные банки для слива, находящиеся в том же поддоне. Электроды промывают дистиллированной водой из промывалки, подставив под электроды стакан. При обнаружении капель ртути в поддоне или при проливании ртути надо немедленно сообщить лаборанту и преподавателю.
ЛАБОРАТОРНАЯ РАБОТА 3
Определение ионов таллия и цинка при совместном
присутствии методом классической полярографии
Цель работы: 1. Изучить влияние мешающих факторов (растворенный кислород и максимум 1-го рода) на вольтамперную кривую.
2. Провести качественный и количественный полярографический анализ ионов металлов при их совместном присутствии.
А. Регистрация полярограммы кислорода. Удаление растворенного кислорода и максимума 1-го рода
Проведению полярографических измерений мешают растворенный кислород и максимумы 1-го рода. В разбавленных водных растворах солей растворяется приблизительно 2·l0-4 M кислорода. Кислород восстанавливается на ртутном электроде. На первой волне, помимо восстановления кислорода, наблюдается полярографический максимум 1-го рода, особенно ярко выраженный в разбавленных фоновых растворах (рис. 16, кривая 1). Полярографические максимумы мешают обработке полярограмм, поэтому их устраняют введением ПАВ. На полярограмме по-прежнему остаются волны восстановления кислорода (рис. 16, кривая 2).
П
еред регистрацией полярограммы удаляют растворенный кислород. Из щелочных и нейтральных растворов кислород довольно быстро удаляется восстановлением сульфит-ионами (О2 + 2Na2SО3 → 2Na2SО4).
Д
Рис. 16. Полярограммы кислорода на фоне 0,01 М КС1
1 – до прибавления желатина;
2 – после добавления желатина;
3 – полярограмма 0,01 М КС1 после
удаления кислорода с помощью Na2SО3
обавление 0,1 г Na2SО3 в 100 мл раствора достаточно для удаления кислорода в течение 5 мин (рис. 16, кривая 3).
Реагенты и аппаратура
Хлорид калия, 0,01 М раствор.
Желатин, 0,1%-ный раствор.
Сульфит натрия, кристаллический.
Ячейка с ртутным капающим электродом.
1. Полярограф LP-60 готовят к работе согласно описанию прибора. Для проверки работоспособности прибора в измерительную цепь вместо ячейки подключают сопротивление (100 кОм) и, включив развертку постоянного напряжения, регистрируют изменение тока в цепи в интервале потенциалов от 0 до -1,2 В (скорость развертки 200 мВ/мин, чувствительность 1/200).
Если прибор исправен, самописец регистрирует прямую линию с наклоном (угол 450) при диапазоне тока (чувствительности) 1/200.
2. Выполнение работы. Электролизер тщательно промывают дистиллированной водой (все сливы собирают в специальную склянку!) и помещают в него 0,01М KCI. Ртутный капающий электрод опускает в ячейку и вынимает из ячейки только преподаватель или лаборант! Кончик капилляра должен быть обязательно погружен в раствор.
Записывают классическую полярограмму (скорость развертки потенциала 200 мВ/мин, чувствительность 1/100) от 0 до -1,5 В.
Ознакомившись с полярограммой (она должна быть похожа на приведенную на рис. 16 кривую 1), убеждаются, что максимум 1-го рода можно устранить с помощью желатина. Для этого добавляют в электролизер 2 капли раствора желатина и записывают полярограмму. Убедившись, что максимум исчез, и на полярограмме присутствуют две волны восстановления кислорода (рис. 16, кривая 2), приступают к удалению растворенного кислорода.
К раствору в электролизере прибавляют примерно 0,1 г Na2SО3 (половина шпателя), перемешивают и через 5 мин записывают полярограмму. Сравнивают полярограмму (она должна быть похожа на приведенную на рис. 16 кривую 3) с предыдущими и убеждаются, что кислород из раствора удален.
Б. Регистрация и расшифровка полярограммы смеси деполяризаторов (полярографический спектр)
Реагенты и аппаратура
Стандартные 0,01 М растворы Т1(1), Zn(II).
0,1 М раствор KCl.
Сульфит натрия, кристаллический.
Желатин, 0,1%-ный раствор.
Полярограф LP-60.
Ячейка с ртутным капающим
электродом.
-
Выполнение работы. К полученному у преподавателя раствору, содержащему ионы Т1(I) и Zn(II), прибавляют примерно 0,1 г сульфита натрия, 2 капли раствора желатины и разбавляют до метки фоновым электролитом (0,1 М KCl).
Электролизер тщательно промывают сначала водой, затем анализируемым раствором, чтобы избежать разбавления. Помещают в электролизер исследуемый раствор так, чтобы кончик капилляра был погружен в раствор.
Записывают классическую полярограмму при непрерывном режиме регистрации тока от -0,2 В до -1,3 В при скорости развертки потенциала 200 мВ/мин (чувствительность 1/100).
Для расшифровки полярограмм очень важно отметить начало записи полярограммы и величину начального потенциала.
2. Расшифровка полярограмм и представление результатов:
а) измеряют высоты всех волн (мм) на полярограмме, проведя касательные ко всем трем участкам волны и опустив вертикаль из верхней точки пересечения на нижнюю касательную;
б) по способу стандартов рассчитывают концентрации ионов таллия и цинка в анализируемом растворе;
в) измеряют величины Е1/2 всех волн с учетом величины начального потенциала и масштаба диаграммы по оси потенциалов.
Найденные значения Е1/2 сравнивают с табличными для 0,1 М раствора KCl и определяют, какие деполяризаторы находятся в растворе.
ЛАБОРАТОРНАЯ РАБОТА 4
Определение малых концентраций ионов кадмия
на осциллографическом полярографе
Цель работы: определение малых концентраций ионов кадмия в растворе методом осциллографической полярографии.
Перед началом работы необходимо ознакомиться с описанием прибора и принципом его работы. После этого приступить к выполнению работы.
Реагенты и аппаратура
Стандартный 10-3 М раствор Cd(II).
0,1 М раствор KCl.
Сульфит натрия, кристаллический.
Желатин, 0,1%-ный раствор.
Полярограф ОП-102.
Ячейка с ртутным капающим
электродом.
Выполнение работы
1. В электролизер (анод – донная ртуть) поместить стандартный 1·10-5 М раствор хлористого кадмия, предварительно подготовленный для полярографирования. Установить ручки управления в соответствующие положения. Зарегистрировать полярограмму, измерить высоту пика, величину тока пика, потенциал пика.
2. В мерную колбу на 50 мл получить у преподавателя раствор соли кадмия и подготовить его для полярографирования, как и стандартный раствор. Электролизер сполоснуть исследуемым раствором.
3. Поместить в электролизер исследуемый раствор. Снять осциллограмму, измерить высоту пика, определить потенциал пика.
4. Рассчитать концентрацию кадмия в анализируемом растворе по способу стандартов.
3. ПОТЕНЦИОМЕТРИЯ
Потенциометрия широко применяется как для определения различных физико-химических величин, так и для определения концентрации веществ в растворах. При этом различают: прямую (ионометрию) и косвенную (потенциометрическое титрование) потенциометрию.
Ионометрия объединяет методы прямого определения концентрации или активности ионов в различных фазах с использованием ионселективных электродов. Ионометрия включает в себя рН-метрию, а также катионо- и анионометрию.
При потенциометрическом титровании в ходе химической реакции наблюдают за изменением потенциала индикаторного электрода. При этом строится интегральная кривая титрования в координатах Е = f (V), по которой определяют точку эквивалентности. Точность потенциометрического титрования возрастает, если определение точки эквивалентности проводят по дифференциальной кривой, построенной в координатах:
Зависимость потенциала индикаторного электрода от активности определяемого компонента описывается уравнением Нернста:
где Е – потенциал индикаторного электрода, В;
E0 – стандартный электродный потенциал, В;
R – универсальная газовая постоянная, равная 8,314 Дж/моль∙К;
Т – температура в градусах Кельвина;
n – число электронов, участвующих в процессе;
F – число Фарадея, равное 96 500 Кл;
а – активность определяемого иона (моль/л).
Если учесть, что измерения проводятся при 25 ºС, и от натурального логарифма перейти к десятичному, то уравнение Нернста будет иметь следующий вид:
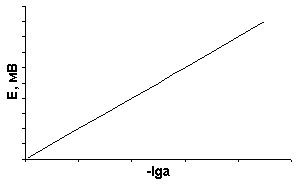
Зависимость потенциала индикаторного электрода от логарифма активности определяемого иона носит линейный характер (рис. 17).
Д
Рис. 17. Зависимость потенциала
электрода от логарифма активности
иона
ля проведения потенциометрических измерений используют два электрода: индикаторный и сравнения.
Потенциал индикаторных электродов прямо или косвенно зависит от концентрации определяемых ионов. Индикаторными являются электроды 1-го рода (например, медная пластинка, помещенная в раствор соли меди) и мембранные (ионселективные), например, стеклянный электрод.
Электроды сравнения. Относят к электродам 2-го рода. Они представляют собой металлическую пластинку, покрытую нерастворимой солью этого металла и погруженную в насыщенный раствор соли с одноименным анионом. Например, хлорсеребряный электрод – это серебряная проволока, покрытая хлоридом серебра и помещенная в насыщенный раствор хлорида калия. Схематическая запись такого электрода выглядит следующим образом:
АgАgClKСlнасыщ.
В некоторых случаях в качестве электрода сравнения используют ртутно-оксидный электрод:
НgНg2ОKОНнасыщ.
Основным требованием к электродам сравнения является постоянство их потенциала. Каломельный электрод представляет собой металлическую ртуть, покрытую слоем каломели, помещенную в насыщенный раствор хлорида калия:
HgHg2Cl2KСlнасыщ.
Потенциал этого электрода зависит от концентрации ионов ртути:
Концентрацию ионов ртути выражают из произведения растворимости каломели:
Поскольку значение ПР является величиной постоянной для данного соединения, концентрация хлорид-ионов поддерживается на одном уровне, то и концентрация ионов ртути также не меняется. Следовательно, и потенциал каломельного электрода остается постоянным при данной температуре.
Потенциометрический метод применим к различным типам химических реакций: нейтрализации, окисления-восстановления, осаждения и комплексообразования. Для использования той или иной химической реакции необходимы:
а) достаточная скорость и быстрое установление равновесия;
б) отсутствие побочных процессов;
в) стехиометричность;
г) возможность выбора соответствующего индикаторного электрода.
Индикаторный электрод выбирается в зависимости от того, какая реакция лежит в основе титрования. Чаще всего составляются следующие пары электродов.
Метод нейтрализации: индикаторный электрод – стеклянный; электрод сравнения – хлорсеребряный или каломельный.
Метод окисления-восстановления: индикаторный электрод – платиновый; электрод сравнения – каломельный.
Методы осаждения и комплексообразования: индикаторный электрод – металлическая проволока из металла, концентрацию ионов которого необходимо определить; электрод сравнения – каломельный.
3.1. Прямые методы потенциометрии. рН-метрия
Методы прямой потенциометрии основаны на непосредственном применении уравнения Нернста для нахождения активности или концентрации иона по экспериментально измеренной ЭДС цепи или потенциалу электрода.
Электроды, применяемые для измерения рН-растворов
Стандартный водородный электрод представляет собой платинированную платиновую пластинку, погруженную в 1 М раствор кислоты, через который пропускают газообразный водород под давлением 1 атм. Схематически этот полуэлемент записывают следующим образом:
Pt, H2 │ 2H+
На поверхности водородного электрода протекает обратимая реакция:
2Н+ + 2е ↔ Н2
Обратимый потенциал водородного электрода определяется соотношением:
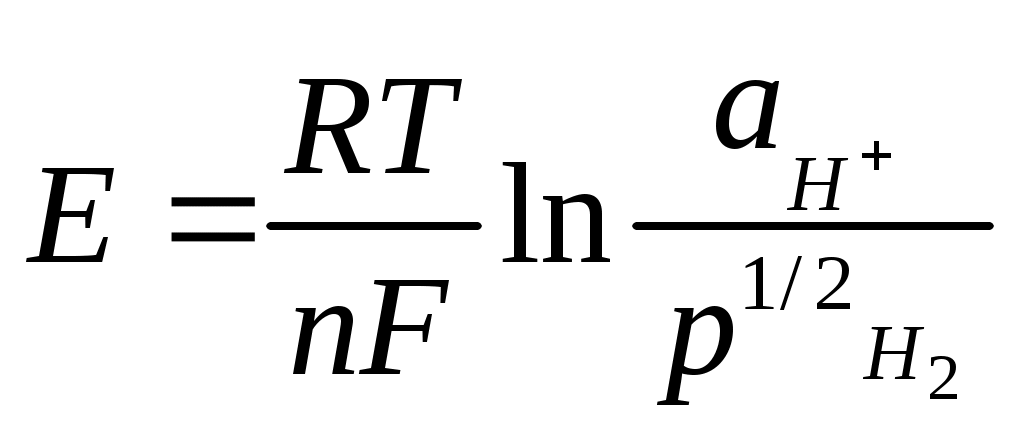 ,
,где р1/2 – парциальное давление водорода, кПа.
По международному соглашению IUPAC потенциал стандартного водородного электрода принят равным 0 В. Электрод применяют редко, поскольку на его потенциал оказывают влияние различные окислители и восстановители. Данный электрод используется для стандартизации буферных растворов.
С
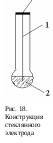 теклянный электрод – это мембранный ионселективный электрод. Он является самым надежным и широко применяемым для измерения рН-растворов, как водных, так и некоторых неводных.
теклянный электрод – это мембранный ионселективный электрод. Он является самым надежным и широко применяемым для измерения рН-растворов, как водных, так и некоторых неводных.Он выполнен в виде тонкостенного шарика, в который вводят хлорсеребряный электрод (1) и 0,1 н раствор HCl (2) (рис. 18). Для измерения рН-раствора составляют следующую цепь:
Ag│AgCl│0,1 M HCl││стекло││анализир. раствор│AgCl│Ag
Как показал метод рентгеноструктурного анализа, стекло представляет собой сетку, построенную из кремнийкислородных цепочек (рис. 19). Пустоты в кремнийкислородном скелете заняты катионами щелочных металлов, которые удерживаются электростатическими полями соседних ионов кислорода.
К
 атионы, находящиеся в пустотах решетки, могут обратимо замещаться на ионы водорода без нарушения структуры решетки. Особенностью состава стекла такого электрода является повышенное содержание оксида щелочного металла (Li2O, Na2O).
атионы, находящиеся в пустотах решетки, могут обратимо замещаться на ионы водорода без нарушения структуры решетки. Особенностью состава стекла такого электрода является повышенное содержание оксида щелочного металла (Li2O, Na2O).Скачок потенциала возникает на поверхности раздела между тонкой стеклянной мембраной и раствором. При этом электрический ток в стекле проводят не электроны, а ионы, следовательно, стекло – это проводник 2-го рода.
Перед работой стеклянный электрод вымачивают в 0,1 н растворе НCl примерно сутки, а затем в течение двух суток отмывают его в дистиллированной воде, постоянно ее меняя. При вымачивании электрода в соляной кислоте поверхностный слой стекла теряет щелочные катионы и насыщается ионами водорода, приобретая водородную функцию:
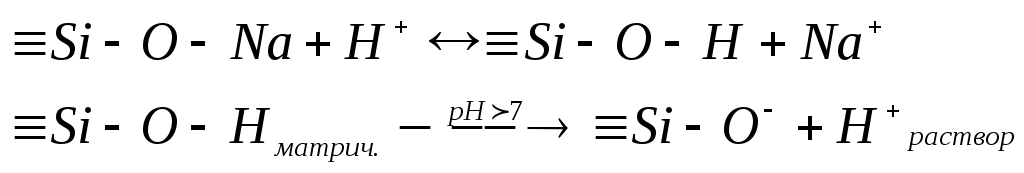
В щелочной среде ионы водорода переходят в раствор, и матрица заряжается отрицательно, а приэлектродный слой – положительно. Возникает двойной электрический слой и соответствующий скачок потенциала. В кислой среде протекают обратные процессы.
З
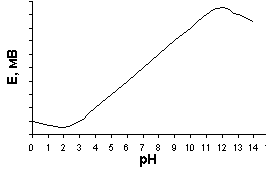
ависимость потенциала стеклянного электрода от рН показана на рис. 20. Данная зависимость характеризуется наличием максимума и минимума. «Щелочная ошибка» проявляется при рН > 12 и связана с тем, что ионы щелочного металла, находящиеся в большом избытке, замещают ионы водорода в стекле. Стекло теряет водородную функцию, одновременно приобретая металлическую.
Н
Рис. 20. Зависимость стеклянного
электрода от рН
аличие минимума на данной зависимости характеризуется «кислотной ошибкой», природа которой до сих пор детально не выяснена.
Потенциал стеклянного электрода представляет собой разность потенциалов на обеих сторонах стеклянной мембраны. Если бы обе стороны мембраны были абсолютно идентичны и активности ионов водорода по обе стороны мембраны одинаковы, то разность потенциалов в данной цепи должна равняться нулю. Однако вследствие разной площади внутренней и внешней поверхностей стекла наблюдается некоторый потенциал (потенциал асимметрии), отличный от нуля, который не учитывается в косвенной потенциометрии в отличие от прямой. Для этого стеклянный электрод калибруют по буферным растворам с известным значением рН.
3.2. Косвенные методы потенциометрии.
Потенциометрическое титрование
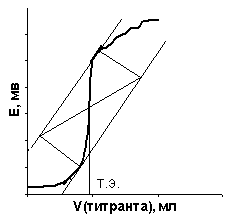
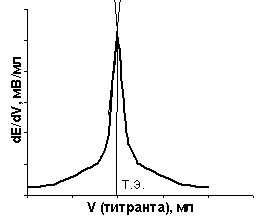
| Рис. 21, а. Интегральная кривая потенциометрического титрования | Рис. 21, б. Дифференциальная кривая потенциометрического титрования |
Потенциометрическое титрование основано на определении точки эквивалентности по результатам измерений. По данным титрования строят либо интегральную зависимость потенциала индикаторного электрода от объема титранта – E = f(V) (рис. 21, а), либо дифференциальную – производная потенциала по объему от объема титранта – ΔE/ΔV = f(V) (рис. 21, б).
Скачок потенциала на кривых потенциометрического титрования зависит от тех же параметров, что и в классическом титровании.
Если в растворе находятся две кислоты различной силы, то первой будет нейтрализована более сильная, а затем – более слабая. На кривой титрования наблюдается два перегиба (скачка): первый соответствует сильной кислоте, второй – слабой (рис. 22).
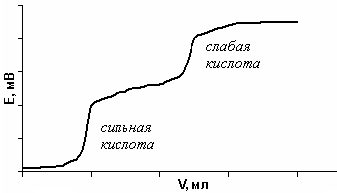
Рис. 22. Кривая потенциометрического титрования слабой
и сильной кислот при совместном присутствии
Если константы диссоциации кислот близки или менее 10-8, то используют различные неводные растворители, которые изменяют силу кислот и оснований.
ЛАБОРАТОРНАЯ РАБОТА 5
Потенциометрическое определение соляной и уксусной кислот при совместном присутствии
Цель работы: количественное определение соляной и уксусной кислот в их смеси методом потенциометрического титрования в среде ацетон–вода.
Количественное определение соляной и уксусной кислот при совместном присутствии в водном растворе осложняется их взаимным влиянием. Кривая титрования характеризуется размытым вторым скачком титрования, что затрудняет нахождение точки эквивалентности.
Хорошо выраженные скачки на кривой титрования можно получить при проведении титрования в среде неводного растворителя, который, обладая дифференцирующим эффектом, изменяет силу кислот. Ацетон, являясь апротонным растворителем, обладает низкой диэлектрической проницаемостью (εвода= 81, εацетона= 20) и проявляет дифференцирующее действие по отношению к кислотам.
В среде ацетона значительно изменяются константы диссоциации соляной и уксусной кислот. В среде ацетона КHCI = 1∙10-4 , KCH3COOH = = 4∙10-13 (в воде KCH3COOH = 1,8 ∙10-5). Отношение констант КHCI/КCH3COOH = 1∙10-4 /4∙10-13 = 2,5∙108 и показывает принципиальную возможность раздельного определения кислот в их смеси.
Порядок работы
1. Ознакомиться с установкой, подготовить установку к работе, включить потенциометр.
2. Получить данные потенциометрического титрования соляной кислоты едким натром в водном растворе.
3. Получить данные потенциометрического титрования уксусной кислоты едким натром в водном растворе.
4. Получить данные потенциометрического титрования смеси соляной и уксусной кислот едким натром в вводно-ацетоновой среде (1:1).
5. Построить интегральные кривые потенциометрического титрования. Построить дифференциальную кривую титрования смеси кислот. Определить точки эквивалентности и рассчитать через титр по определяемому веществу количество уксусной и соляной кислот в индивидуальных растворах и в смеси (г/мл).
Реагенты и аппаратура