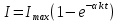Добавлен: 23.11.2023
Просмотров: 144
Скачиваний: 3
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
 с), когда триплетный уровень не заселен, интенсивность флуоресценции достигает максимального значения. Затем за время соизмеримое со временем жизни триплетных молекул она уменьшается и достигает стационарного значения. Этот процесс соответствует переходу части молекул в триплетное состояние.
с), когда триплетный уровень не заселен, интенсивность флуоресценции достигает максимального значения. Затем за время соизмеримое со временем жизни триплетных молекул она уменьшается и достигает стационарного значения. Этот процесс соответствует переходу части молекул в триплетное состояние. Интенсивность флуоресценции по мере заселения триплетного уровня уменьшается со временем по экспоненциальному закону. Это уменьшение обусловлено заселением триплетного уровня и насколько эффективно заселяется триплетный уровень, настолько сильно уменьшается интенсивность флуоресценции. Вследствие этого, заселенность триплетного уровня можно рассчитать следующим образом:
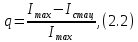
где
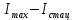 характеризует число молекул перешедших в триплетное состояние,
характеризует число молекул перешедших в триплетное состояние, 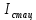 – интенсивность флуоресценции в стационарном состоянии,
– интенсивность флуоресценции в стационарном состоянии, 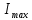 – максимальная интенсивность флуоресценции пропорциональная общему числу молекул в растворе, участвующих в излучении.
– максимальная интенсивность флуоресценции пропорциональная общему числу молекул в растворе, участвующих в излучении.В том случае, когда триплетный уровень не заселяется или заселяется очень слабо, интенсивность флуоресценции остается постоянной равной
и она зависит от интенсивности возбуждения линейно. Регистрацию спектров флуоресценции удобнее вести в стационарном режиме, однако это приводит к неправильному определению интенсивности флуоресценции, так как зависит от мощности возбуждения. В процессе эксперимента, вследствие нестабильности работы лампы или эффекта внутреннего фильтра, может произойти изменение стационарной интенсивности флуоресценции, а вследствие этого изменение заселенности триплетного уровня молекул. При этом максимальная интенсивность флуоресценции будет оставаться постоянной. Для определения заселенности триплетного уровня трифенилена вторая методика оказалась более удобной. Однако контроль осуществляли с использованием первой методики. Совпадение результатов обеих методик позволило сделать вывод о правомерности использования обеих методик.
При определении относительной заселенности триплетного и синглетного уровней молекул основной вклад в ошибку вносит случайная ошибка, возникающая при измерении времен разгорания и затухания фосфоресценции. Величина погрешности при определении времени разгорания и затухания фосфоресценции в секундном диапазоне обуславливалась флуктуациями фототока, нелинейностью усилителя, погрешностью блока временной развертки и механической постоянной графопостроителя. Три последних источника по данным многократных проверок могли дать в сумме систематическую ошибку не более 1%. Для уменьшения влияния фототока измерения повторялись 5-10 раз и случайная ошибка в каждом конкретном случае находилась с использованием коэффициентов Стьюдента при доверительной вероятности 0,90.
Определенная, с учетом сказанного, абсолютная ошибка при измерении относительной заселенности триплетного уровня молекул равнялась 0,02 единицы.
3. Влияние фотопревращений примесных центров на формирование квазилинейчатых спектров люминесценции
3.1. Фотопреврашения ПАУ в системах Шпольского
Бытует мнение, что полициклические ароматические углеводороды не испытывают фотопревращений при ультрафиолетовом облучении. Но обнаружение и исследование фотохимических превращений ПАУ представляет огромный интерес по нескольким причинам. Во-первых, они могут привести к уменьшению энергии возбуждения, что повлечет за собой переход части упорядоченной энергии электронного возбуждения в неупорядоченную энергию[57]. Во-вторых, реакции фотопревращения можно использоваться для «выжигания» в спектре поглощения замороженных растворов устойчивых провалов[58], распространенный метод для решения ряда задач спектроскопии [59,60]. Одними из первых фотохимические превращения для некоторых ПАУ были обнаружены в ряде работ [61], что говорит о необходимости исследования данного вопроса. Фотопревращения молекул ПАУ могут привести к ошибкам в спектрофлуориметрическом анализе органических соединений по квазилинейчатым спектрам, в том случае, если будет образовываться устойчивый люминесцирующий продукт.
Фотопревращение трифенилена в отвердевшем растворе метилциклогексана впервые было описано в работе. Спектры поглощения и флуоресценции были отнесены к виду дигидротррифенилена, возникающие при внедрении единичной молекулы растворителя к ароматическому углеводороду. Функция создания фотопродукта состоит из поочередного поглощения пары фотонов, однако, при этом нижнее триплетное состояние является промежуточным. Также в работе была определена суммарная эффективность реакции поглощения второго фотона, которая составила примерно
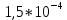 на длине волны 265 нм, а в видимой области в 1000 раз меньше (
на длине волны 265 нм, а в видимой области в 1000 раз меньше (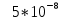 ), что обусловлено сильным триплетным поглощением в этой области. По словам авторов при ультрафиолетовом облучении ртутной лампой формируется флуоресцирующий фотопродукт. При длительном воздействии на образец возрастает интенсивность свечения фотопродукта. В работе так же указано, что данная реакция не происходит в температурном диапазоне от 19 до 26
), что обусловлено сильным триплетным поглощением в этой области. По словам авторов при ультрафиолетовом облучении ртутной лампой формируется флуоресцирующий фотопродукт. При длительном воздействии на образец возрастает интенсивность свечения фотопродукта. В работе так же указано, что данная реакция не происходит в температурном диапазоне от 19 до 26  (комнатная температура). Следовательно, свойства различны – при низких температурах возрастает время жизни триплетного состояния, возрастает квантовый выход фотореакции, происходящей в данном состоянии, в результате возрастания степени заселенности триплетного уровня молекул за счет увеличения времени жизни. Исходя из работы [62] более подходящими условиями для проявление двухквантовых реакций являются: возрастание интенсивности возбуждающего света и реакционное время жизни. Двухквантовые реакции проще всего производятся в замороженных растворах ароматических молекул в алифатических растворителях так же в полимерных средах, или же просто в жестких средах. Скорость двухквантовой реакции зависит от квадрата интенсивности возбуждающего света.
(комнатная температура). Следовательно, свойства различны – при низких температурах возрастает время жизни триплетного состояния, возрастает квантовый выход фотореакции, происходящей в данном состоянии, в результате возрастания степени заселенности триплетного уровня молекул за счет увеличения времени жизни. Исходя из работы [62] более подходящими условиями для проявление двухквантовых реакций являются: возрастание интенсивности возбуждающего света и реакционное время жизни. Двухквантовые реакции проще всего производятся в замороженных растворах ароматических молекул в алифатических растворителях так же в полимерных средах, или же просто в жестких средах. Скорость двухквантовой реакции зависит от квадрата интенсивности возбуждающего света. В настоящей работе было изучено фотопревращение трифенилена в матрицах Шпольского при температуре 77 K (в замороженных твердых растворах). Наиболее «удобным» растворителем считается н.-гептан для получения квазилинейчатых спектров, так же в нем получаются наиболее резкие спектры люминесценции трифенилена, чем в иных растворителях [62]. В противоположность н.-гептану н.-декан является «неудобным» растворителем [62], спектры в н.-декане получаются с широкими полосами. В момент записи спектров люминесценции трифенилена возбуждение осуществлялось азотным лазером ЛГИ-21, интенсивностью ослабленной нейтральным фильтром в 3 раза. На рис. 4 представлен без предварительного облучения спектр люминесценции трифенилена в растворителе н.-гептане при 77 K. Спектр люминесценции трифенилена нормирован по отношению к интенсивности линии 352,87 нм. Расположение линий в спектре и распределение интенсивности в спектре не противоречат литературным данным [63].
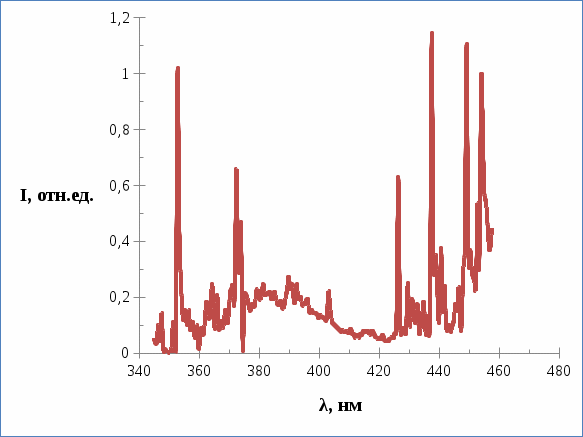
Рис. 4. Спектр люминесценции трифенилена в н.-гептане без предварительного облучения.
Далее исследуемый образец облучался азотным лазером ЛГИ-21 без уменьшения интенсивности в течение 10 минут и снова записывался спектр люминесценции трифенилена, который представлен на рис. 5.
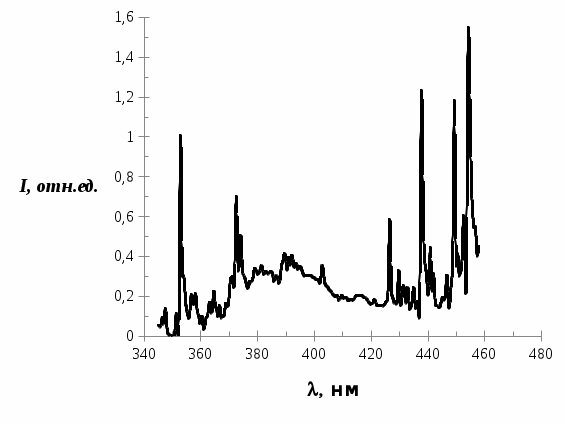
Рис. 5. Спектр люминесценции трифенилена в н.-гептане после облучения в течение 10 минут.
Как видно, в спектре люминесценции трифенилена в области от 360 нм до 420 нм проявляется дополнительное свечение, которое отсутствует в случае образца без облучения. Интенсивность дополнительного свечения возрастает со временем и достигает некого значения.
Дальнейшее облучение образца вплоть до 90 минут привело, к тому что в спектре трифенилена появилось очень сильное свечение, возрастающее со временем, которое можно увидеть на рисунках 6-8.
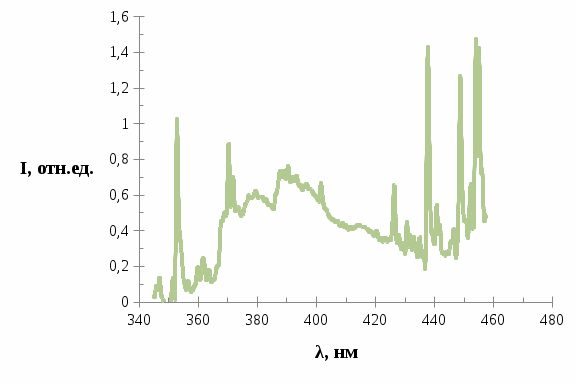
Рис. 6. Спектр люминесценции трифенилена в н.-гептане после облучения в течение 30 минут.
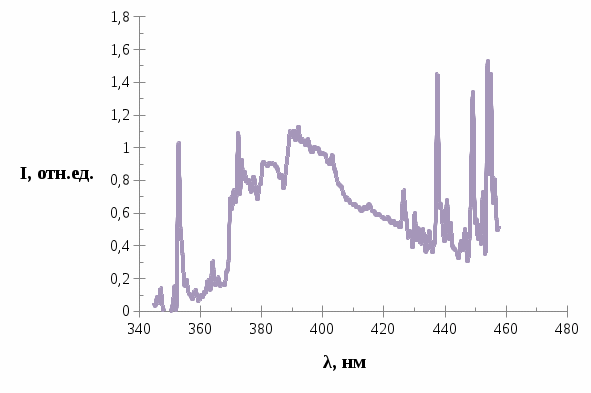
Рис. 7. Спектр люминесценции трифенилена в н.-гептане после облучения в течение 60 минут.
Спектры на рис.4, рис.5, рис.6, рис.7, рис.8 приведены для концентрации трифенилена в растворе
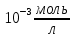 . В спектре проявляются широкие полосы с максимумами в области 370, 380 и 390 нм.
. В спектре проявляются широкие полосы с максимумами в области 370, 380 и 390 нм. 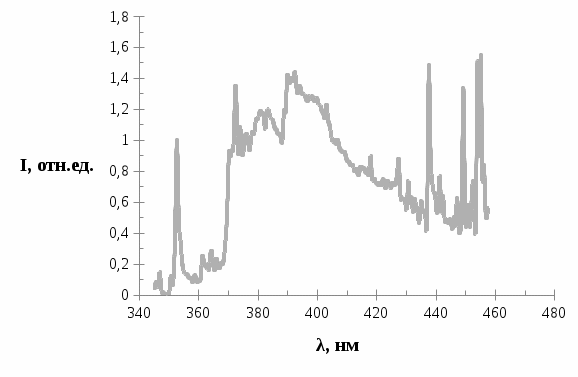
Рис. 8. Спектр люминесценции трифенилена в н.-гептане после облучения в течение 90 минут.
Полученный нами фотопродукт по свои параметрам (распределением интенсивности в спектре и положением полос) совпадает с параметрами спектра фотопродукта трифенилена в метилциклогексане при 77 K [33] При сравнении рис.4 (первоначальный спектр) и рис.8 (при облучении лазером на протяжении полутора часов) видно, что возрастает свечение фотопродукта в 5-6 раз, при этом происходит уменьшение интенсивности фосфоресценции трифенилена всего на 5-10%. Снижение интенсивности квазилинейчатого спектра фосфоресценции трифенилена говорит о том, что за фотопревращение в данном случае, ответственны внедренные молекулы в кристаллы н.-парафина, то есть молекулы трифенилена.
С целью проверки не является ли свечение фотопродукта итогом фотопревращения примесей трифенилена или растворителя (так же воды и кислорода) было выполнено фракционирование трифенилена с помощью хроматографической колонки. В результате фракционирования было выявлено семь фракций. В доказательство теории о вероятности проявления фотопродукта обусловлено фотопревращением трифенилена, а никак не присутствием в составе образца примесей, люминесценция фотопродукта прослеживается только лишь в тех фракциях, в каких присутствует трифенилен.
На рис.9 представлен график, демонстрирующий повышение фотопродукта трифенилена с увеличение времени при облучении лазером ЛГИ-21. Как видно, прослеживается существенное увеличение свечения фотопродукта, который с ходом времени ощущает истощение.
Рассмотрим кинетику изменения числа молекул фотопродукта со временим. Пусть
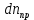 – изменение числа молекул фотопродукта за время dt, тогда с течением времени это число изменяется по закону:
– изменение числа молекул фотопродукта за время dt, тогда с течением времени это число изменяется по закону: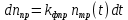 (3.1)
(3.1)где
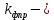 константа фотопревращения, которая характеризует вероятность превращения молекул трифенилена поcле поглощения второго кванта,
константа фотопревращения, которая характеризует вероятность превращения молекул трифенилена поcле поглощения второго кванта,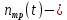 число молекул трифенилена в растворе.
число молекул трифенилена в растворе.В начальный момент времени изменение числа молекул фотопродукта было равно 0:
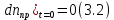
Из одной молекулы трифенилена может образоваться одна молекула фотопродукта, тогда:
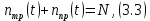
где N
 число молекул трифенилена до начала реакции.
число молекул трифенилена до начала реакции.Подставляя (3.3) в (3.1) получим дифференциальное уравнение, показывающие изменение числа молекул фотопродукта со временем:
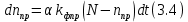
Здесь α
коэффициент, который определяет долю молекул трифенилена, находящихся в триплетном состоянии. Решая это уравнение, получим: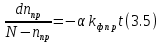
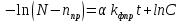 (3.6)
(3.6)С учетом начальных условий, С=N, тогда:
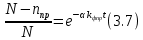
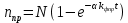 (3.8)
(3.8)Выражение (3.8) справедливо для части молекул трифенилена, которая учувствует в фотореакции. Поскольку интенсивность линий в квазилинейчатых спектрах пропорциональна числу молекул, тогда для интенсивности можно записать: