Добавлен: 24.10.2023
Просмотров: 211
Скачиваний: 11
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Пирамидный синдромразвивается, как правило, на ранней стадии БАС и проявляется оживлением сухожильных рефлексов. Вслед за этим нередко развивается нижний спастический парапарез. В руках повышение рефлексов сочетается с мышечными атрофиями, т.е. наблюдается сочетанное, одновременное поражение центральных (пирамидных) путей и периферического мотонейрона, что является характерным для БАС. Поверхностные брюшные рефлексы исчезают по мере прогрессирования процесса. Симптом Бабинского (при штриховом раздражении подошвы большой палец стопы разгибается, другие пальцы веерообразно расходятся и разгибаются) наблюдается в половине случаев заболевания. Могут быть нарушения чувствительности. У больных наблюдаются парестезии в дистальных отделах рук и ног. Боли, порою выраженные, обычно ночные, могут быть связаны с тугоподвижностью суставов, длительной неподвижностью, спазмами вследствие высокой спастичности, с крампи (болезненными мышечными спазмами), депрессией. Выпадения чувствительности не характерны. Глазодвигательные нарушения не характерны и встречаются на терминальной стадиях заболевания. Нарушения функций тазовых органов не характерны, но в далеко зашедшей стадии может наблюдаться задержка или недержание мочи. Умеренно выраженные когнитивные расстройства(снижение памяти и умственной работоспособности) проявляются у половины больных. У 5% больных развивается деменция лобного типа, которая может сочетаться с паркинсоническим синдромом. Особенностью БАС является отсутствие пролежней даже у парализованных лежачих больных.
Прогрессирование двигательных расстройств заканчивается смертью через несколько (2—6) лет. Иногда болезнь имеет острое течение.
Дифференциальная диагностика
Для дифференциации бокового амиотрофического склероза от потенциально излечимых и/или имеющих доброкачественный прогноз заболеваний проводится МРТ позвоночника и головного мозга. С ее помощью выявляют признаки дегенерации пирамидных трактов, которые характерны для пирамидного и классического вариантов БАС.
Кроме того, в связи со схожими симптомами и клинической картиной боковой амиотрофический склероз необходимо дифференцировать от:
заболеваний мышц (миозит с клеточными отклонениями, дистрофическая миотония Россолимо-Штейнерта-Куршмана, окулофаренгиальная миодистрофия);
заболеваний с поражением нервно-мышечного синапса (миастения, синдром Ламберта-Итона);
заболеваний периферических нервов (мультифокальная моторная невропатия с блоками проведения, синдром Персонейджа-Тернера, изолированные моторные полиневропатии, проксимальная диабетическая моторная полиневропатия, нейромиотония Исаакса);
заболеваний спинного мозга (бульбоспинальная амиотрофия Кеннеди, а также иные спинальные амиотрофии взрослых, хроническая вертеброгенная ишемическая миелопатия, сирингомиелия, опухоли спинного мозга, семейная спастическая параплегия, дефицит гексозаминидазы, хронический лимфолейкоз или лимфома с поражением периферических мотонейронов);
заболеваний головного мозга (дисциркуляторная энцефалопатия, мультисистемная атрофия, сирингобульбия, опухоли задней черепной ямки и краниоспинального перехода);
системных заболеваний.
ДИАГНОСТИКА
Диагностика бокового амиотрофического склероза в первую очередь базируется на тщательном анализе клинической картины заболевания. ЭМГ исследование (электромиография) подтверждает диагноз болезни мотонейрона.
Боковой амиотрофический склероз нужно заподозрить:
•при развитии слабости и атрофий, а возможно и фасцикуляций (мышечных подергиваний) в мышцах кисти
•при похудания мышц тенара одной из кистей с развитием слабости аддукции (приведения) и оппозиции большого пальца (обычно асимметрично)
•при этом наблюдается затруднение при схватывании большим и указательным пальцами, затруднения при подбирании мелких предметов, при застегивании пуговиц, при письме
•при развитии слабости в проксимальных отделах рук и плечевом поясе, атрофий в мышцах ног в сочетании с нижним спастическим парапарезом
•при развитии у пациента дизартрии (нарушений речи) и дисфагии (нарушений глотания)
•при появлении у пациента крампи (болезненных мышечных сокращений)
Диагностические критерии БАС всемирной организации неврологов (1998):
•поражение (дегенерация) нижнего мотонейрона, доказанное клинически, электрофизиологически или морфологически
•поражение (дегенерация) верхнего мотонейрона по данным клинической картины
•прогрессирующее развитие субъективных и объективных признаков заболевания на одном уровне поражения центральной нервной системы или распространение их на другие уровни, определяемое по данным анамнеза или обследования.
Подозрение на БАС: •при наличии симптомов поражения нижнего мотонейрона в 2 или 3 регионах, таких как прогрессирующая мышечная атрофия или другие двигательные симптомы
ЭМГ(электромиография)
Характерные изменения и находки на ЭМГ при БАС:
•фибрилляции и фасцикуляции в мышцах верхних и нижних конечностей, или в конечностях и области головы
•уменьшение количества двигательных единиц и увеличение амплитуды и длительности потенциала действия двигательных единиц
•нормальная скорость проведения в нервах, иннервирующих мало пораженные мышцы, и снижения скорости проведения в нервах, иннервирующих тяжело пораженные мышцы (скорость должна быть не менее 70% от нормальной величины)
•нормальная электрическая возбудимость и скорость проведения импульса по волокнам чувствительных нервов.
Лечение:
Цели терапии:
• замедлить прогрессирование болезни и продлить период заболевания, при котором больной не нуждается в постоянном постороннем уходе. • уменьшить выраженность отдельных симптомов болезни и поддерживать стабильный уровень жизни.
Лекарственная терапия:
Единственный препарат, достоверно замедляющий прогрессирование БАС, РИЛУЗОЛ (пресинаптический ингибитор высвобождения глутамата)
Препарат продлевает жизнь больных в среднем на 3 месяца.
Метаболическая терапия:
- витамин Е
- витамины группы В
- АТФ
- ноотропы
- кортексин
- Анаболические гормоны
- L - картинин
- глицин
Рекомендуют введение трипептида (тиреотропин-релизинг-гормон) в больших дозах внутривенно или в малых дозах подкожно и внутримышечно. Возможно также применение дериватов этого гормона.
Для улучшения нервно-мышечной проводимости назначают дибазол, прозерин. Для уменьшения спастичности – мидокалм.
Лечение проводят курсами по несколько раз в год.
Возможное альтернативное лечение
1) Трансплантация стволовых клеток (пуповинная кровь человека) -
медленное прогрессирование заболевания в 70% исследованных случаев. Доказанные преимущества – в течение 3 месяцев замедление прогрессирования заболевания,сохранение остаточной силы в конечностях.
Прогноз:
Прогноз всегда неблагоприятный, за исключением редких наследственных случаев, ассоциирующихся с определенными мутациями в гене супероксиддисмутазы-1. Длительность болезни при бульбарном дебюте в среднем составляет 2, 5 года. При спинальном 3, 5 года. Лишь 7% больных живут дольше 5 лет.
Список литературы:
1.Боковой амиотрофический склероз/ под ред., И.А. Завалишин. – М.: ГЭОТАР-Медиа, 2009.
2.Дифференциальная диагностика в неврологии и нейрохирургии/ под ред., Е.И. Гусевой. – М.: ГЭОТАР-Медиа, 2005.
3.Неврология: национальное руководство/ под ред., Е.И. Гусевой, А.Н. Коновалова, В.И. Скворцовой, А.Б. Гехт – М.: ГЭОТАР-Медиа, 2009.
Приложение А.

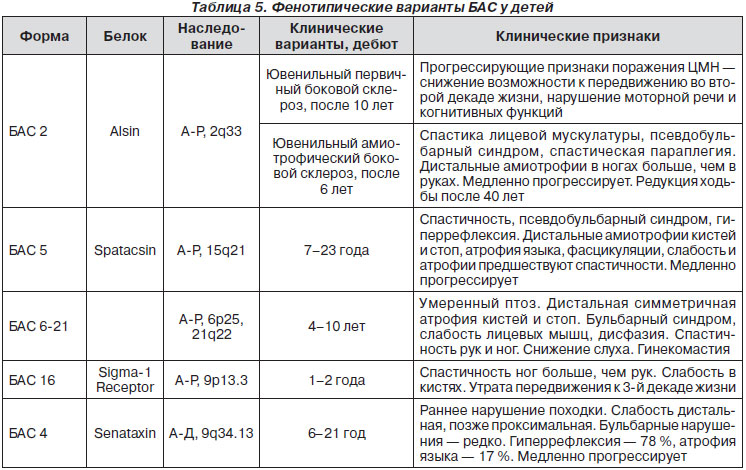

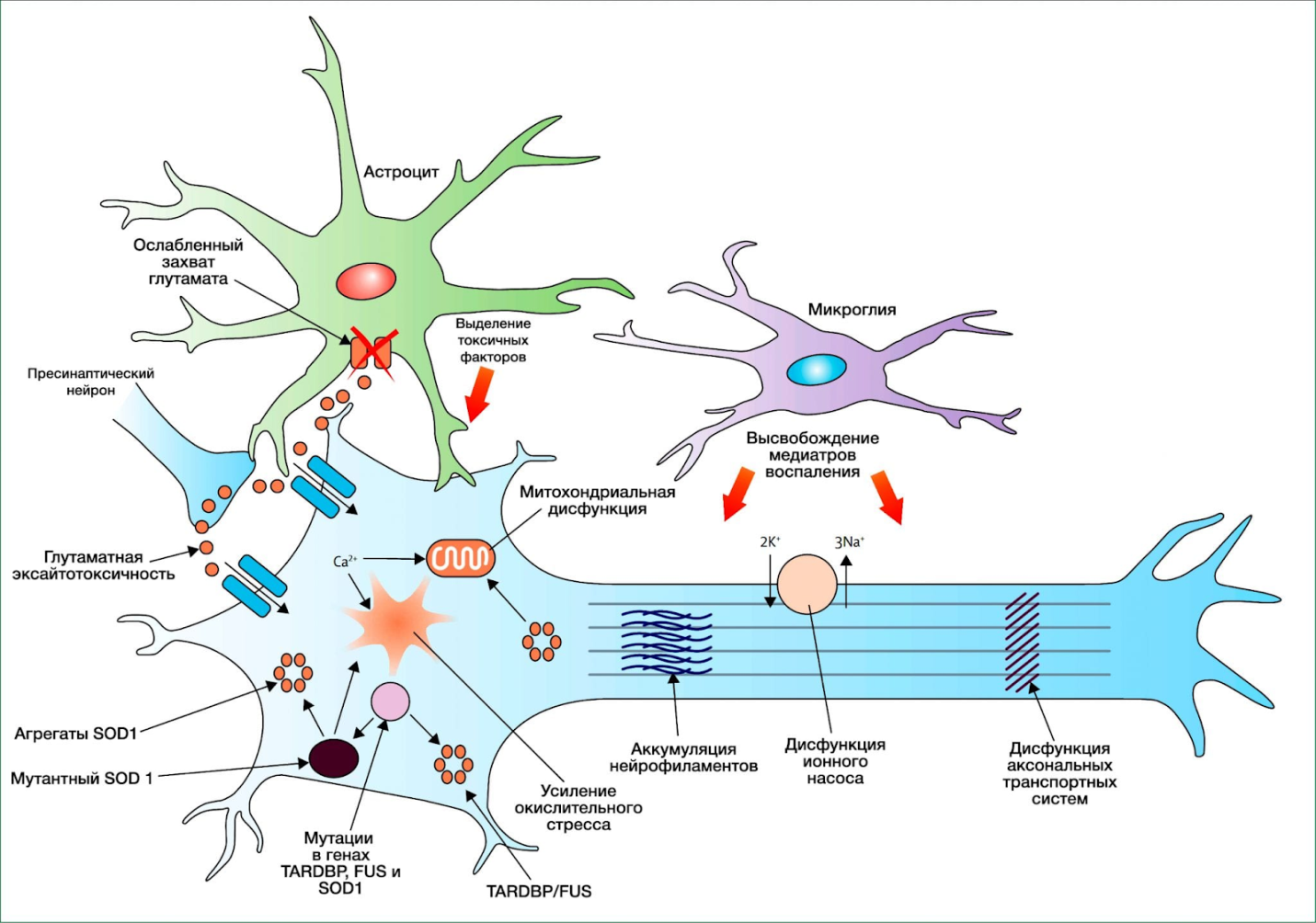
Прогрессирование двигательных расстройств заканчивается смертью через несколько (2—6) лет. Иногда болезнь имеет острое течение.
Дифференциальная диагностика
Для дифференциации бокового амиотрофического склероза от потенциально излечимых и/или имеющих доброкачественный прогноз заболеваний проводится МРТ позвоночника и головного мозга. С ее помощью выявляют признаки дегенерации пирамидных трактов, которые характерны для пирамидного и классического вариантов БАС.
Кроме того, в связи со схожими симптомами и клинической картиной боковой амиотрофический склероз необходимо дифференцировать от:
заболеваний мышц (миозит с клеточными отклонениями, дистрофическая миотония Россолимо-Штейнерта-Куршмана, окулофаренгиальная миодистрофия);
заболеваний с поражением нервно-мышечного синапса (миастения, синдром Ламберта-Итона);
заболеваний периферических нервов (мультифокальная моторная невропатия с блоками проведения, синдром Персонейджа-Тернера, изолированные моторные полиневропатии, проксимальная диабетическая моторная полиневропатия, нейромиотония Исаакса);
заболеваний спинного мозга (бульбоспинальная амиотрофия Кеннеди, а также иные спинальные амиотрофии взрослых, хроническая вертеброгенная ишемическая миелопатия, сирингомиелия, опухоли спинного мозга, семейная спастическая параплегия, дефицит гексозаминидазы, хронический лимфолейкоз или лимфома с поражением периферических мотонейронов);
заболеваний головного мозга (дисциркуляторная энцефалопатия, мультисистемная атрофия, сирингобульбия, опухоли задней черепной ямки и краниоспинального перехода);
системных заболеваний.
ДИАГНОСТИКА
Диагностика бокового амиотрофического склероза в первую очередь базируется на тщательном анализе клинической картины заболевания. ЭМГ исследование (электромиография) подтверждает диагноз болезни мотонейрона.
Боковой амиотрофический склероз нужно заподозрить:
•при развитии слабости и атрофий, а возможно и фасцикуляций (мышечных подергиваний) в мышцах кисти
•при похудания мышц тенара одной из кистей с развитием слабости аддукции (приведения) и оппозиции большого пальца (обычно асимметрично)
•при этом наблюдается затруднение при схватывании большим и указательным пальцами, затруднения при подбирании мелких предметов, при застегивании пуговиц, при письме
•при развитии слабости в проксимальных отделах рук и плечевом поясе, атрофий в мышцах ног в сочетании с нижним спастическим парапарезом
•при развитии у пациента дизартрии (нарушений речи) и дисфагии (нарушений глотания)
•при появлении у пациента крампи (болезненных мышечных сокращений)
Диагностические критерии БАС всемирной организации неврологов (1998):
•поражение (дегенерация) нижнего мотонейрона, доказанное клинически, электрофизиологически или морфологически
•поражение (дегенерация) верхнего мотонейрона по данным клинической картины
•прогрессирующее развитие субъективных и объективных признаков заболевания на одном уровне поражения центральной нервной системы или распространение их на другие уровни, определяемое по данным анамнеза или обследования.
Подозрение на БАС: •при наличии симптомов поражения нижнего мотонейрона в 2 или 3 регионах, таких как прогрессирующая мышечная атрофия или другие двигательные симптомы
ЭМГ(электромиография)
Характерные изменения и находки на ЭМГ при БАС:
•фибрилляции и фасцикуляции в мышцах верхних и нижних конечностей, или в конечностях и области головы
•уменьшение количества двигательных единиц и увеличение амплитуды и длительности потенциала действия двигательных единиц
•нормальная скорость проведения в нервах, иннервирующих мало пораженные мышцы, и снижения скорости проведения в нервах, иннервирующих тяжело пораженные мышцы (скорость должна быть не менее 70% от нормальной величины)
•нормальная электрическая возбудимость и скорость проведения импульса по волокнам чувствительных нервов.
Лечение:
Цели терапии:
• замедлить прогрессирование болезни и продлить период заболевания, при котором больной не нуждается в постоянном постороннем уходе. • уменьшить выраженность отдельных симптомов болезни и поддерживать стабильный уровень жизни.
Лекарственная терапия:
Единственный препарат, достоверно замедляющий прогрессирование БАС, РИЛУЗОЛ (пресинаптический ингибитор высвобождения глутамата)
Препарат продлевает жизнь больных в среднем на 3 месяца.
Метаболическая терапия:
- витамин Е
- витамины группы В
- АТФ
- ноотропы
- кортексин
- Анаболические гормоны
- L - картинин
- глицин
Рекомендуют введение трипептида (тиреотропин-релизинг-гормон) в больших дозах внутривенно или в малых дозах подкожно и внутримышечно. Возможно также применение дериватов этого гормона.
Для улучшения нервно-мышечной проводимости назначают дибазол, прозерин. Для уменьшения спастичности – мидокалм.
Лечение проводят курсами по несколько раз в год.
Возможное альтернативное лечение
1) Трансплантация стволовых клеток (пуповинная кровь человека) -
медленное прогрессирование заболевания в 70% исследованных случаев. Доказанные преимущества – в течение 3 месяцев замедление прогрессирования заболевания,сохранение остаточной силы в конечностях.
Прогноз:
Прогноз всегда неблагоприятный, за исключением редких наследственных случаев, ассоциирующихся с определенными мутациями в гене супероксиддисмутазы-1. Длительность болезни при бульбарном дебюте в среднем составляет 2, 5 года. При спинальном 3, 5 года. Лишь 7% больных живут дольше 5 лет.
Список литературы:
1.Боковой амиотрофический склероз/ под ред., И.А. Завалишин. – М.: ГЭОТАР-Медиа, 2009.
2.Дифференциальная диагностика в неврологии и нейрохирургии/ под ред., Е.И. Гусевой. – М.: ГЭОТАР-Медиа, 2005.
3.Неврология: национальное руководство/ под ред., Е.И. Гусевой, А.Н. Коновалова, В.И. Скворцовой, А.Б. Гехт – М.: ГЭОТАР-Медиа, 2009.
Приложение А.