Файл: Взаимодействие генов и их проявление при разных типах наследования.docx
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 25.10.2023
Просмотров: 256
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Биохимические методы.
Биохимические методы основаны на изучении активности ферментных систем и позволяют выявить наследственные заболевания, обусловленные генными мутациями. С помощью биохимических методов описано более 1000 врожденных болезней обмена ве-ществ (это дефекты структурных, транспортных и др. белков).
Применение данных методов в пренатальном периоде или непосредственно после рождения позволяет своевременно выявить патологию и начать лечение, как, например, в случае фенилкетонурии.
ж) Методы дерматоглифики и пальмоскопии.
В 1892 году Ф.Гальтон установил, что кожные гребешковые узоры пальцев и ладоней являются индивидуальной характеристикой человека и не меняются в течении всей его жизни. Этот метод сейчас широко используется в криминалистике и генетических исследованиях (например, установление зиготности близнецов установления отцовства и некоторых анеуплодий).
Популяционно-статистический метод.
Этим методом можно рассчитать частоту встречаемости в популяции различных аллелей гена и разных генотипов по этим аллелям, выяснить распространение в ней различных наследственных, признаков, в том числе и патологических. Основной для выяснение генетической структуры популяции является закон Харди-Вайнберга.
и) Иммуногенетические методы.
Применяются при обследовании на иммунодефицитные состояния, при подозрении на антигенную несовместимость матери и плода, при установлении истинного отцовства, а также определения наследственного предрасположения к болезням. Так, например, HLA-типирование используется для пренатальной диагностики адреногенитального синдрома (врожденной гиперплазии коры надпочечников).
к) Экспресс-методы.
Экспресс методы - это быстрые предварительные методы изучения генетики человека. Они часто используются для исследования больших контингентов людей с целью выявления наследственной патологии как скрининг-методы, применяемые при проведении просеивающих программ. Например, скрининг новорожденных на фенилкетонурию, беременных на альфафетоироин, при помощи которого можно пренатально определить у плода некоторые пороки развития (например, анэнцефалию, открытые формы спинно-мозговых грыж, синдром Дауна).
Так к экспресс-методам относятся:
Выявление Х-хроматина, который осуществляется посредством соскоба клеток слизистой оболочки щеки из которых делают мазок, окрашивают его и просматривают препарат в обычном микроскопе. Метод позволяет определить количество Х-хромосом в кариотипе по количеству телец Барра.
Методы пренатальной диагностики.
Пренатальная диагностика связана с решением биологических и этических проблем до рождения ребенка, т.к. речь идет о предупроеждении рождения ребенка с патологией, не поддающейся лечению. На современном уровне развития пренатальной диагностики можно установить диагноз всех хромосомных болезней, большинства врожденных поро-ков развития, энзимопатий, при которых известен биохимимческий дефект. Часть из них можно установить практически на любом сроке беременности (хромосомные болезни), часть после 12-й недели (редукционные пороки конечностей, анэнцефалию), часть только во второй половине беременности (пороки сердца, почек).
В настоящее время применяют прямые (исследуют плод) и непрямые (исследуют беременную) методы. К прямым неинвазивным (без хирургического вмешательства) ме-тодам относятся ультрасонография (УЗИ). К прямым инвазивным (с нарушением це-лостности тканей) - хорионбиопсяи, амниоцентез и фетоскопия.
При хорионбиопсии производится взятие эпителия ворсинок хориона через канал шейки матки под контролем УЗИ между 8-й и 10-й неделями беременности.
В результате амниоцентеза получают амниотическую жидкость и клетки плода для последующего анализа. Пункцию проводят в начале второго триместра беременности (15-17 неделя) через брюшину под контролем УЗИ.
Материал полученный при хорионбиопсии и амниоцентезе используют для биохимических исследований (генные мутации), а клетки - для анализа ДНК (хромосомные и геномные мутации). Осложнения при этом методе исследования не превышает 1%.
Фенилкетонурия (ФКУ)
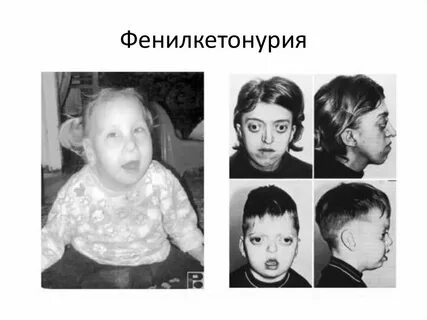
-
Фенилкетонурия -
Проявления: светлая кожа, шаткая походка, тремор рук и ног, судорожные припадки, наличие у больного мышиного запаха -
Аутосомно-рецесс -
биохимическое исследование мочи
Альбинизм.

Мелатонин не вырабатывается
2.Проявляется отсутствием нормальной для данного вида окраски кожи, волос, шерсти, радужной и пигментной оболочек глаз
3.Аут-рец наследование (нормальный аллель (вариант) подавляет проявление мутантного аллеля (варианта), т.е. мутация может проявиться, только находясь в гомозиготном состоянии. При этом мутантный ген расположен в аутосоме (неполовой хромосоме) и
наследование не сцеплено с полом.)
4.Посмотреть на пациента
Метод диагностики: дерматоглифика.? Биохимический? Применяется в криминалистике и генетических исследованиях
Галактоземия
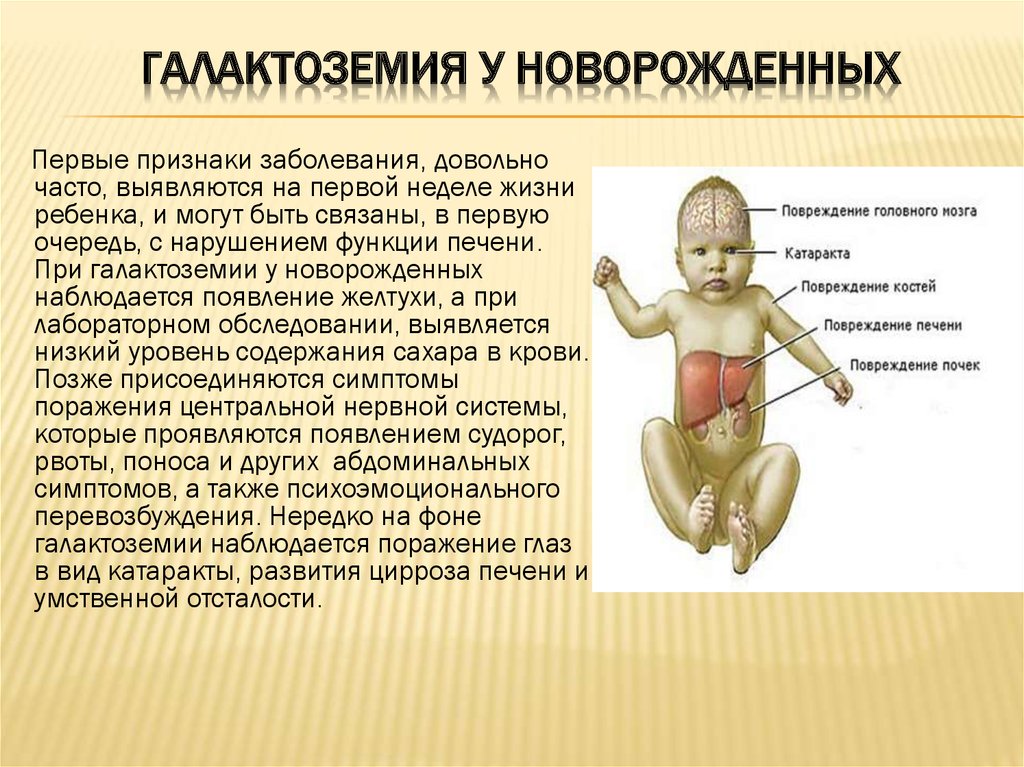

-
Галактоземия -
Основными симптомами заболевания являются: желтуха новорожденных, рвота, понос с обезвоживанием организма, умственное недоразвитие, гепатоспленомегалия (увеличение печени и селезенки) с циррозом. -
аутосомно-рецессивному -
Диагноз ставится с помощью биохимических методов: на основании обнаружения галактозы в крови и моче, а также галактозо-1-фосфата в крови.
Болезнь Тея-Сакса.

-
Болезнь Тея-Сакса -
Заболевание проявляется на первом году жизни, чаще в возрасте 4-6 месяцев в виде резкого отставания психического развития, снижение зрения с последующей слепотой, дегенерацией интеллекта до идиотии, судорожных припадков, параличей. На глазном дне атрофия сосков зрительных нервов и вишнево-красное пятно в макулярной области. Большинство детей умирают в возрасте до 2-х лет -
Аутосомно-рецессивный -
Диагноз ставится на основании клинических проявлений, генеалогического анализа, определения в крови вакуолизированных лейкоцитов. Возможна пренатальная диагностика.
Синдром Марфана
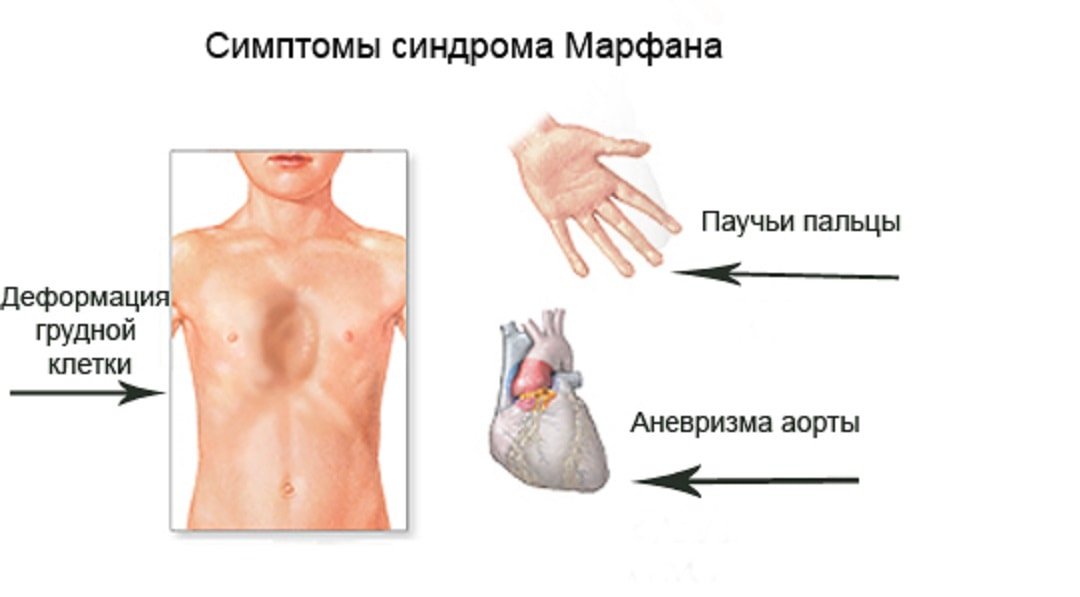

3. Тип наследования синдрома — аутосомно-доминантный.
4
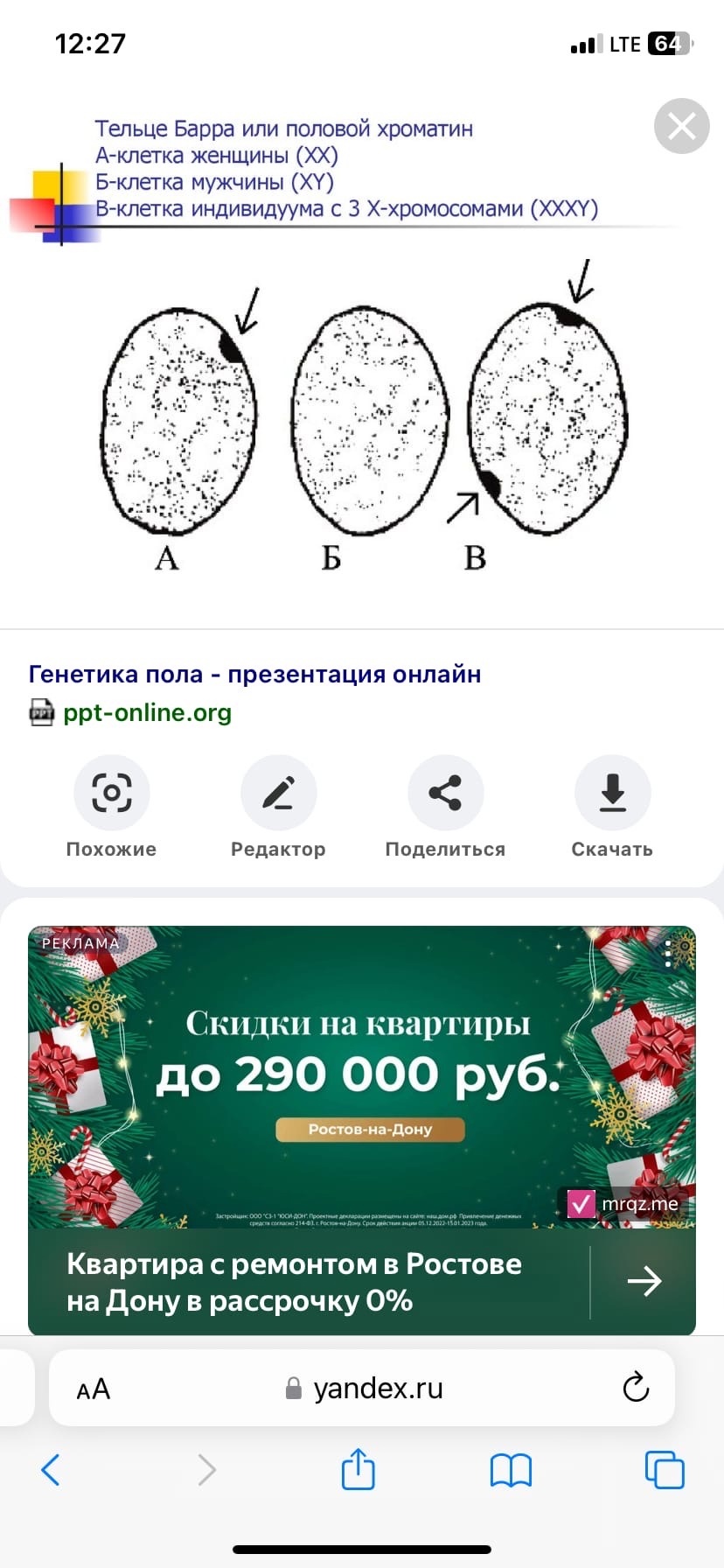 .генетическая диагностика
.генетическая диагностикаХРОМОСОМНЫЕ БОЛЕЗНИ В ХОДЕ ОНТОГЕНЕЗА. РАСЧЕТ ГЕНЕТИЧЕСКОГО РИСКА
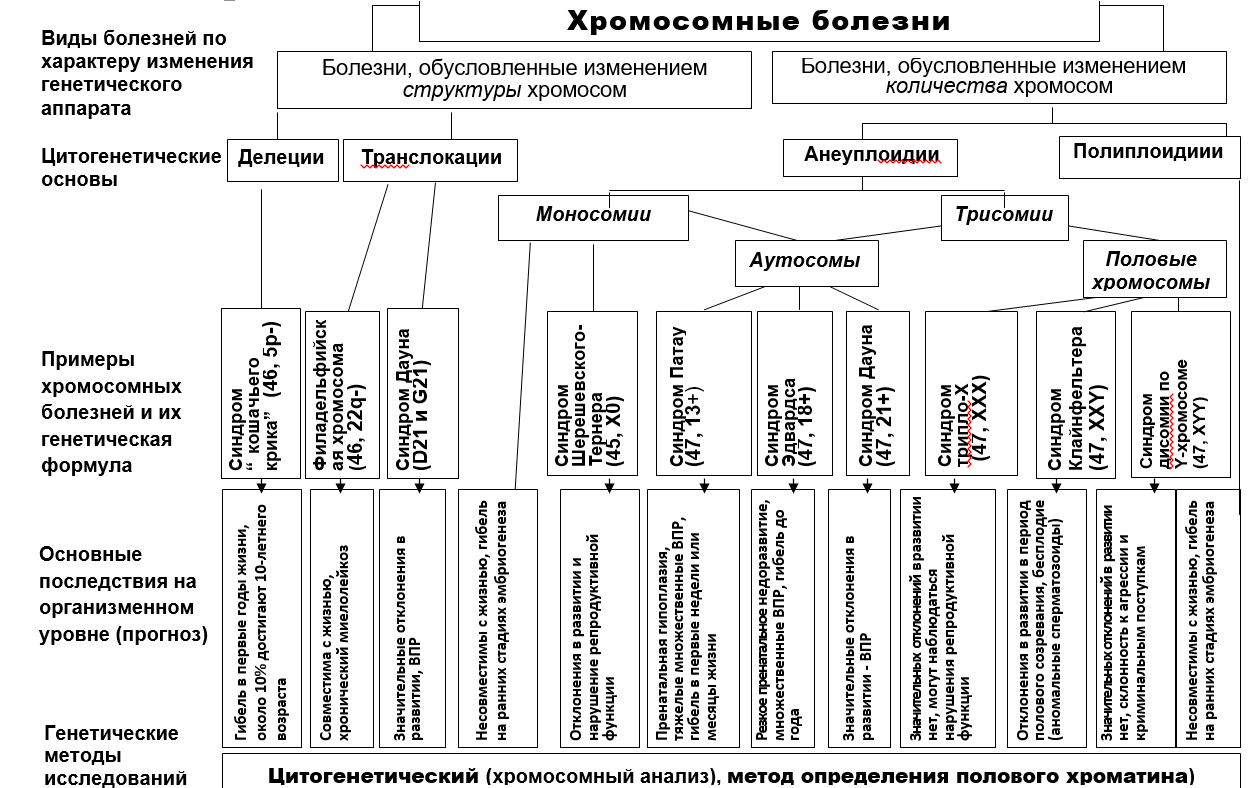
Синдром кошачьего крика (синдром Лежена)

-
Синдром Кошачьего крика (синдром Лежена) -
характерный плач ребенка; изменение формы головы; недоразвитие нижней челюсти;
низкий вес тела;
дефекты развития пальцев;
косолапость. отставание в умственном развитии;
пониженный мышечный тонус;
нарушение координации движений;
запоры;
лунообразное лицо;
короткая шея;
лабильность поведения; проблемы со зрением. В возрасте 18 – 25 лет, до которого доживает менее 5% детей, на первый план выходит отставание в умственном развитии. Больной не может выполнять какую-либо работу. При мозаичной форме болезни способность к обучению несколько лучше. У таких пациентов есть шанс быть интегрированными в общество. Внешность больных характеризуется теми же врожденными нарушениями, которые были описаны при рождении. Иногда наблюдается ускоренное старение кожи.
-
Кариотип 46 XX или XY, 5р- (короткого плеча) делеция -
Проведение инвазивной пренатальной диагностики (амниоцентеза, биопсии ворсин хориона или кордоцентеза) и непосредственного анализа генетического материала плода.
После рождения предварительный диагноз синдрома «кошачьего крика» устанавливается неонатологом на основании типичных диагностических признаков (характерного плача, фенотипических черт, множественных стигм дизэмбриогенеза). Для подтверждения хромосомной патологии проводится цитогенетическое исследование.
Механизм возникновения делеция
Синдром Патау.

-
Синдром Патау -
У детей с синдромом Патау присутствуют тяжелые врожденные пороки развития головного мозга, черепа, внутренних органов, опорно-двигательного аппарата.
Характерные признаки заболевания:
неправильно развитые кости лицевого и мозгового черепа;
измененное строение стоп, кистей;
симметричная многопалость;
укорочение шеи;
близкое расположение глазниц;
сужение глазных щелей;
отсутствие радужной оболочки, глазного яблока;
расщелина губы, неба;
деформация, неправильное расположение ушных раковин;
западание переносицы и др.
У младенцев с синдромом Патау недоразвиты основные мозговые структуры центральной нервной системы, пищеварительная система. Тяжелые пороки развития наблюдаются в сердечно-сосудистой системе – дефекты крупных кровеносных сосудов, сердечных перегородок. Страдает мочеполовая система: двурогая матка, крипторхизм (неопущение яичек), гипоплазия (недоразвитие) наружных половых органов. 47, 13+
-
Первый этап пренатального скрининга включает в себя ультразвуковое исследование, определение биохимических маркеров (бета-ХГЧ, РАРР-А и др.).
Беременным, которые попали в группу риска, показаны следующие методы инвазивной пренатальной диагностики:
в 8–12 недель – биопсия ворсин хориона;
в 14–18 недель – амниоцентез;
в 20 недель и больше – кордоцентез.
Полученный материал исследуют на наличие трисомии хромосомы 13 методом КФ-ПЦР, кариотипированием с дифференциальной окраской хромосом.