ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 06.07.2024
Просмотров: 124
Скачиваний: 0
СОДЕРЖАНИЕ
1.Классификация и номенклатура ферментов
4) Структура белковой молекулы
8)Биосинтез холерестерина и его биологическая роль.
10) .Роль желчных кислот, переваривание липидов
11) Характеристика гормонов поджелудочной железы
15) Характеристика витаминов(в2 и в5)
19)Ферменты биологического окисления
20)Взаимосвязь аминоуглеводов с обменом липидов
23) Факторы, влияющие на скорость ферментативных реакций
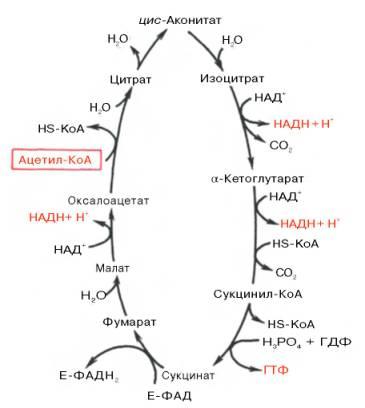
35)Цикл Кребса
|
Цикл трикарбоновых кислотвпервые был открыт английским биохимиком Г. Кребсом. Он первым постулировал значение данного цикла для полного сгорания пирувата, главным источником которого является гликолитическое превращение углеводов. В дальнейшем было показано, что цикл трикарбо-новыхкислотявляется тем центром, в котором сходятся практически все метаболические пути. Таким образом,цикл Кребса– общий конечный путьокисленияацетильныхгрупп (в виде ацетил-КоА), в которые превращается в процессекатаболизмабольшая часть органическихмолекул, играющих роль «клеточноготоплива»:углеводов,жирных кислотиаминокислот. Образовавшийся в результате окислительного декарбоксилированияпирувата вмитохондрияхацетил-КоА вступает вцикл Кребса. Данный цикл происходит в матриксемитохондрийи состоит из восьмипоследовательных реакций(рис. 10.9). Начинается цикл с присоединения ацетил-КоА к оксалоацетату и образованиялимонной кислоты(цитрата). Затемлимонная кислота(шестиуглеродное соединение) путем рядадегидрирований(отнятиеводорода) и двухдекарбоксилирований(отщепление СО2) теряет два углеродныхатомаи снова вцикле Кребсапревращается в оксалоацетат (четырехуглеродное соединение), т.е. в результате полного оборота цикла однамолекулаацетил-КоА сгорает до СО2 и Н2О, а молекулаокса-лоацетата регенерируется. Рассмотрим все восемьпоследовательных реакций(этапов)цикла Кребса.
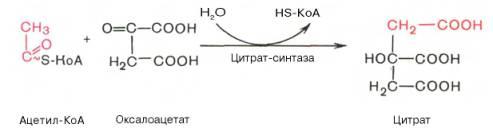
Рис. 10.9. Цикл трикарбоновых кислот(цикл Кребса). Первая реакциякатализируетсяферментомцит-рат-синтазой, при этомацетильнаягруппа ацетил-КоА конденсируется с оксалоацетатом, в результате чего образуетсялимонная кислота:
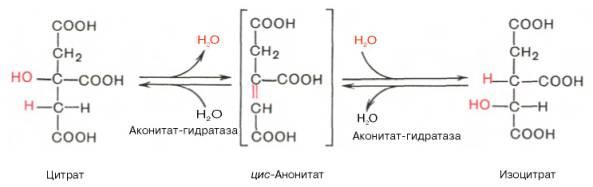
По-видимому, в данной реакциив качестве промежуточного продукта образуется связанный сферментомцитрил-КоА. Затем последний самопроизвольно и необратимо гидролизуется с образованиемцитратаи HS-KoA. В результате второй реакцииобразовавшаясялимонная кислотаподвергается дегидратированию с образованием цис-аконитовойкислоты, которая, присоединяямолекулуводы, переходит визолимонную кислоту(изоцитрат). Катализирует эти обратимыереакциигидратации–дегидратацииферментаконитатгидратаза (аконитаза). В результате происходит взаимоперемещение Н и ОН вмолекулецитрата:
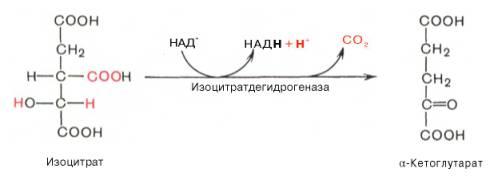
Третья реакция, по-видимому, лимитирует скоростьцикла Кребса.Изолимонная кислотадегидрируется в присутствии НАД-зависимой изо-цитратдегидрогеназы.
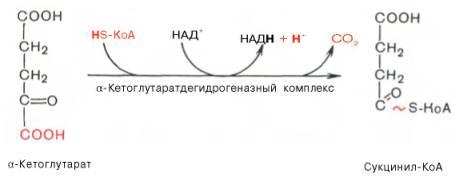
В ходе изоцитратдегидрогеназной реакцииизолимонная кислотаодновременно декарбоксилируется. НАД-зависимая изоцитратдегидрогеназа является аллостерическимферментом, которому в качестве специфическогоактиваторанеобходимАДФ. Кроме того,ферментдля проявления своейактивностинуждается вионахMg2+или Мn2+. Во время четвертой реакциипроисходит окислительное декарбокси-лирование α-кетоглутаровойкислотыс образованием высокоэнергетического соединения сукцинил-КоА. Механизм этойреакциисходен с таковымреакцииокислительногодекарбоксилированияпирувата до ацетил-КоА, α-кетоглутаратдегидрогеназный комплекс напоминает по своей структуре пируватдегидрогеназный комплекс. Как в одном, так и в другом случае вреакциипринимают участие 5коферментов: ТПФ, амидлипоевой кислоты, HS-KoA, ФАД и НАД+.
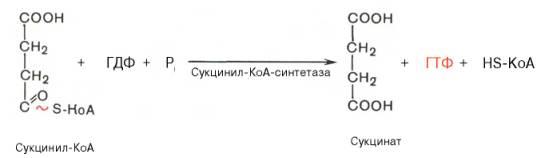
Пятая реакциякатализируетсяферментомсукцинил-КоА-синтета-зой. В ходе этойреакциисукцинил-КоА при участии ГТФ инеорганического фосфатапревращается вянтарную кислоту(сукцинат). Одновременно происходит образование высокоэргической фосфатной связи ГТФ за счет высокоэргической тиоэфирной связи сукцинил-КоА:
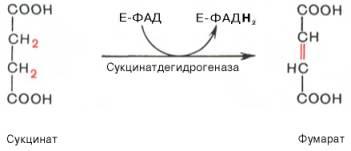
В результате шестой реакциисукцинатдегидрируется вфумаровую кислоту.Окислениесукцинатакатализируетсясукцинатдегидрогеназой, вмолекулекоторой сбелкомпрочно (ковалентно) связанкоферментФАД. В свою очередьсукцинатдегидрогеназапрочно связана с внутренней ми-тохондриальноймембраной:
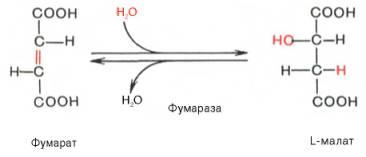
Седьмая реакцияосуществляется под влияниемферментафума-ратгидратазы (фумаразы). Образовавшаяся при этомфумаровая кислотагидратируется, продуктомреакцииявляетсяяблочная кислота(малат). Следует отметить, что фумаратгидратаза обладаетстереоспецифичностью(см. главу 4) – в ходереакцииобразуется L-яблочнаякислота:
Наконец, в ходе восьмой реакциицикла трикарбоновых кислотпод влиянием митохондриальной НАД-зависимоймалатдегидрогеназыпроисходитокислениеL-малата в оксалоацетат:
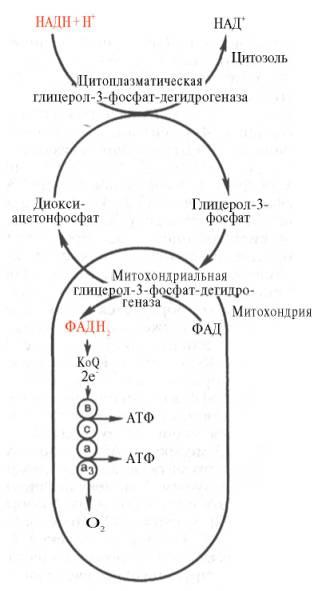
Как видно, за один оборот цикла, состоящего из восьми ферментативных реакций, происходит полноеокисление(«сгорание») одноймолекулыацетил-КоА. Для непрерывной работы цикла необходимо постоянное поступление в систему ацетил-КоА, акоферменты(НАД+ и ФАД), перешедшие в восстановленное состояние, должны снова и снова окисляться. Это окислениеосуществляется в системе переносчиковэлектроноввдыхательной цепи(вцепи дыхательныхферментов), локализованной вмембранемитохондрий. Образовавшийся ФАДН2 прочно связан с СДГ, поэтому он передает атомыводородачерез KoQ. Освобождающаяся в результатеокисленияацетил-КоА энергия в значительной мере сосредоточивается в макроэргических фосфатных связяхАТФ. Из 4паратомовводорода3парыпереносят НАДН на систему транспортаэлектронов; при этом в расчете на каждуюпарув системе биологическогоокисленияобразуется 3молекулыАТФ(в процессе сопряженногоокислительного фосфорилирования), а всего, следовательно, 9молекулАТФ(см. главу 9). Однапараатомовот сукцинатдегидрогеназы-ФАДН2 попадает в систему транспорта электроновчерез KoQ, в результате образуется только 2молекулыАТФ. В ходецикла Кребсасинтезируется также однамолекулаГТФ (субстратноефосфорилирование), что равносильно одноймолекулеАТФ. Итак, приокисленииодноймолекулыацетил-КоА вцикле Кребсаи системеокислительного фосфорилированияможет образоваться 12молекулАТФ. Если подсчитать полный энергетический эффект гликолитического расщепления глюкозыи последующегоокислениядвух образовавшихсямолекулпирувата до СО2 и Н2О, то он окажется значительно большим. Как отмечалось, одна молекулаНАДН (3молекулыАТФ) образуется при окислительномдекарбоксилированиипирувата в ацетил-КоА. При расщеплении одноймолекулыглюкозыобразуется 2молекулыпирувата, а приокисленииих до 2молекулацетил-КоА и последующих 2 оборотовцикла трикарбоновых кислотсинтезируется 30молекулАТФ(следовательно,окислениемолекулыпирувата до СО2 и Н2О дает 15 молекулАТФ). К этому количеству надо добавить 2молекулыАТФ, образующиеся при аэробномгликолизе, и 6молекулАТФ, синтезирующихся за счетокисления2молекулвнемитохондриального НАДН, которые образуются приокислении2молекулглицеральдегид-3-фосфата в дегидрогеназнойреакциигликолиза. Следовательно, при расщеплении втканяходноймолекулыглюкозыпо уравнению С6Н12О6 + 6О2 —> 6СО2 + 6Н2О синтезируется 38 молекулАТФ. Несомненно, что в энергетическом отношении полное расщеплениеглюкозыявляется более эффективным процессом, чем анаэробныйгликолиз. Необходимо отметить, что образовавшиеся в процессе превращения глицеральдегид-3-фосфата 2 молекулыНАДН в дальнейшем приокислениимогут давать не 6молекулАТФ, а только 4. Дело в том, что самимолекулывнемитохондриального НАДН не способны проникать черезмембранувнутрьмитохондрий. Однако отдаваемые имиэлектронымогут включаться в митохондриальную цепь биологическогоокисленияс помощью так называемого глицеролфосфатного челночного механизма (рис. 10.10). Ци-топлазматический НАДН сначала реагирует с цитоплазматическим ди-гидроксиацетонфосфатом, образуя глицерол-3-фосфат.Реакциякатализи-
Рис. 10.10. Глицеролфосфатный челночный механизм. Объяснение в тексте. руется НАД-зависимой цитоплазматической глицерол-3-фосфат-дегидроге-назой: Дигидроксиацетонфосфат + НАДН + Н+ <=> Глицерол-3-фосфат + НАД+. Образовавшийся глицерол-3-фосфат легко проникает через митохонд-риальную мембрану. Внутримитохондриидругая (митохондриальная) глицерол-3-фосфат-дегидрогеназа (флавиновыйфермент) снова окисляет глицерол-3-фосфат до диоксиацетонфосфата: Глицерол-3-фосфат + ФАД <=> Диоксиацетонфосфат + ФАДН2. Восстановленный флавопротеин(фермент-ФАДН2) вводит на уровне KoQ приобретенные им электроныв цепь биологическогоокисленияи сопряженного с нимокислительного фосфорилирования, а диоксиаце-тонфосфат выходит измитохондрийвцитоплазмуи может вновь взаимодействовать с цитоплазматическим НАДН + Н+. Таким образом, параэлектронов(из одноймолекулыцитоплазматического НАДН + Н+), вводимая вдыхательную цепьс помощью глицеролфосфатного челночного механизма, дает не 3, а 2АТФ.
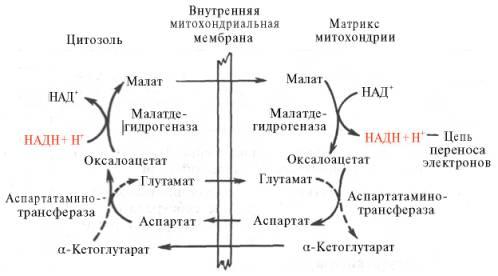
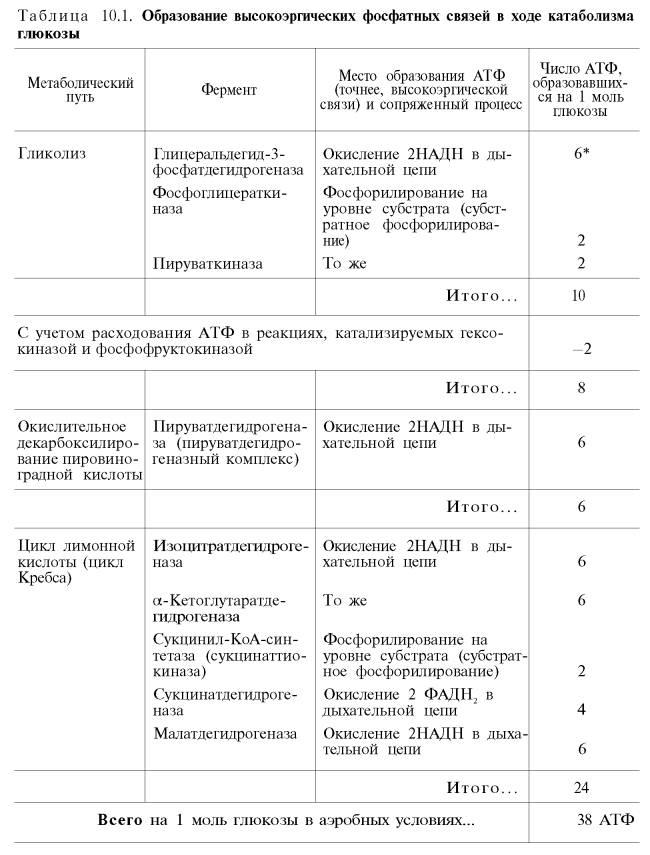
Рис. 10.11. Малат-аспартатная челночная система для переноса восстанавливающих эквивалентов от цитозольного НАДН в митохондриальный матрикс. Объяснение в тексте. В дальнейшем было показано, что с помощью данного челночного механизма лишь в скелетных мышцах и мозге осуществляется перенос восстановленных эквивалентов от цитозольного НАДН + Н+ в митохондрии. В клеткахпечени, почек и сердца действует более сложная малат-ас-партатная челночная система. Действие такого челночного механизма становится возможным благодаря присутствиюмалатдегидрогеназыи ас-партатаминотрансферазы как в цитозоле, так и вмитохондриях. Установлено, что от цитозольного НАДН + Н+ восстановленные эквиваленты сначала при участии ферментамалатдегидрогеназы(рис. 10.11) переносятся на цитозольный оксалоацетат. В результате образуется малат, который с помощью системы, транспортирующейдикарбоновые кислоты, проходит через внутреннююмембранумитохондриив матрикс. Здесь малат окисляется в оксалоацетат, а матриксный НАД+восстанавливается в НАДН + Н+, который может теперь передавать свои электронывцепь дыхательныхферментов, локализованную на внутреннеймембранемитохондрии. В свою очередь образовавшийся оксалоацетат в присутствии глутамата иферментаАсАТ вступает вреакциютрансаминирования. Образующиеся аспарат и α-кетоглутарат с помощью специальных транспортных систем способны проходить черезмембранумитохондрий. Транспортирование в цитозоле регенерирует оксалоацетат, что вызывает к действию следующий цикл. В целом процесс включает легкообратимые реакции, происходит без потребления энергии, «движущей силой» его является постоянноевосстановлениеНАД+ в цитозоле гли-церальдегид-3-фосфатом, образующимся прикатаболизмеглюкозы. Итак, если функционирует малат-аспартатный механизм, то в результате полного окисленияодноймолекулыглюкозыможет образоваться не 36, а 38молекулАТФ(табл. 10.1).
В табл. 10.1 приведены реакции, в которых происходит образование высокоэргических фосфатных связей в ходекатаболизмаглюкозы, с указанием эффективности процесса в аэробных и анаэробных условиях. 36)Гормоны регулирующие обмен углеводов Гормон Действие гормона Изменение секреции гормона при мышечной деятельности средней тяжести
Тироксин или тетрайодтиронин Усиливает процессы окисления жиров, углеводов и белков в клетках, ускоряя, таким образом, обмен веществ в организме. Повышает возбудимость центральной нервной системы. Практически не меняется.
Инсулин Облегчает проникание сахара из крови в клетки мышц и жировой ткани, облегчает проникновение аминокислот из крови в клетки, способствует синтезу белка и жиров. Способствует отложению глюкозы в запас (в печени). В начале работы - увеличивается, облегчая проникновение глюкозы в клетки, а затем - снижается, так как вызывает изменения, противоположные тем, которые необходимы для эффективной мышечной деятельности.
Глюкагон Оказывает действие, во многом противоположное инсулину. Усиливает распад цепочек глюкозы в клетках и выход глюкозы из мест ее хранения в кровь. Стимулирует распад жира в жировой ткани. Увеличивается, обеспечивая распад и выход в кровь углеводов и жиров, дающих энергию для мышечного сокращения. Тироксин или тетрайодтиронин Усиливает процессы окисления жиров, углеводов и белков в клетках, ускоряя, таким образом, обмен веществ в организме. Повышает возбудимость центральной нервной системы. Практически не меняется.
Трийодтиронин Действие во многом аналогично тироксину. Практически не меняется.
Тирокальцитонин Регулирует обмен кальция в организме, снижая его содержание в крови, и увеличивая его содержание в костной ткани (оказывает действие, обратное паратгормону паращитовидных желез). Снижение уровня кальция в крови уменьшает возбудимость центральной нервной системы. Повышается при значительном утомлении, наступающем при выполнении длительной мышечной деятельности. 37) Энергетика аэробного превращения глюкозы. При аэробном превращении глюкозы в конечном счете образуется углекислый газ и вода. Энергетика -38 молекул АТФ. Условно аэробное превращение глюкозы можно разбить на 3 основых этапа:
38) Роль аминокислот в биосинтезе фосфолипидов Принимают участие ам/к-ты: серин и матионин. Начинается биосинтез фосфолипидов так же как 3-глицеридов до образования фосфотивной кислоты.
40)Биологическое окисление( тканевое дыхание) Биологическое окисление- совокупность реакций окисления, проникающие в клетки живого организма. Сущность биологического окисления заключается в обеспечении организма энергией, которая будет затрачиваться на реакции синтеза взаимопревращения веществ, мышечную работу, поддержание терморегуляции. Источником энергии являются продукты питания, содержащие пищ. белки, жиры, углеводы. Они попадают в жел/киш тракт, под действием гидролитических фертментов распадаются на более простые соед. которые далее попадаютв клетки тканей. В каждой клетке находятся сложные ферментные системы: В-окисление Цикл Кребса Гликолиз С помощью которых поступившие в клетку вещества видоизменяются с уелью приобретения форм более доступных для окисления. Сам процесс оксиления под действием молекул кислорода получ. название бтол. окисление. При окислении углеводов и липидов в конечном итоге образуется СО2 и Н2О. При окислении продуктов гидролиза белков, т.е ам/к-т кроме СО2 и Н2О образ-ся аммиак NH3,мочевина и мочевая кислота, т.к при окислении белков, жиров и углеводов вне организма (при горении) , обр-ся практически те же конеч. продукты, как при биол. окислении кроме мочевины и м.к-ты) поэтому биологическое окисление с давних пор сравнивают с горением. Отличия:
90% всех связей энергии, которая образуется при биологическом окислении запас. в фосфатных связях АТФ.
|



42)механизм действия ферментов. Структура и функции ферментов, а также механизм их действия почти ежегодно подробно обсуждаются на многих международных симпозиумах и конгрессах. Важное место отводится рассмотрению структуры всеймолекулыферментаи ееактивных центров, молекулярному механизму действия различных типовферментов, общей теории энзиматическогокатализа. Тем не менее досихпор нет полной ясности по двум кардинальным проблемам энзимологии: чем вызваныспецифичностьдействия и высокая каталитическая эффективностьферментов?
До установления химической природы ферментовгипотезы о механизме их действия опирались на исследования кинетики и модельные опыты химическогогомогенного катализа. Повышениескорости химических реакцийпод действиемферментовобъясняли следующим: а) активированиемсубстратав результате образования адсорбционных или молекулярных, обратимо диссоциирующих фермент-субстратных комплексов; б) цепныммеханизмом реакцийс участием радикалов или возбужденныхмолекул. Оказалось, что цепныемеханизмы реакциине играют существенной роли в биологическомкатализе. После установления химическойприроды ферментовподтвердилось представление, выдвинутое более 80 лет назад В. Анри, Л. Михаэлисом и М. Ментен, о том, что при энзиматическомкатализеферментЕ соединяется (в принципе обратимо) со своимсубстратомS, образуя нестойкий промежуточный фермент-субстратный комплекс ES, который в концереакциираспадается с освобождениемферментаи продуктовреакцииР. Благодаря высокому сродству связывания и образованию ES-комплекса резко возрастает числомолекулсубстрата, вступающих вреакции. Эти представления легли в основу теории «ключа-замка» Э. Фишера, которую иногда называют теорией «жесткойматрицы». Таким образом, жесткая структураактивного центраоказывается комплементарной молекулярной структуресубстрата, обеспечивая тем самым высокуюспецифичностьфермента.
Л. Михаэлис не только постулировал образование промежуточного фермент-субстратного ES-комплекса, но и рассчитал влияниеконцентрациисубстратанаскорость реакции. В процессереакцииразличают несколько стадий: присоединениемолекулысубстратакферменту, преобразование первичного промежуточного соединения в один или несколько последовательных (переходных) комплексов и протекающее в одну или несколько стадий отделение конечных продуктовреакцииотфермента. Это можно схематически проиллюстрировать следующими примерами:
![]()
В реакцияханаболизма, например А + В —> АВ,ферментможет соединяться как с одним, так и с другимсубстратомили обоимисубстратами:
В реакцияхкатаболизма, например АВ —> А + В:
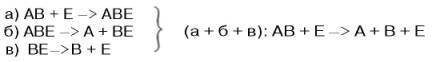
На рис. 4.7 представлена схема образования промежуточного фермент-субстратного комплекса. Еслиферментвактивном центресодержиткофермент, то предполагается образование тройного комплекса (рис. 4.8).
Ферментвступает во взаимодействие ссубстратомна очень короткий период, поэтому долгое время не удавалось показать образование такого комплекса. Прямые доказательства существования фермент-субстратного комплекса были получены в лабораториях Д. Кейлина и Б. Чанса. В настоящее время экспериментальные и математические методы кинетики,термодинамикии статической механикихимических реакцийпозволяют определить для ряда ферментативныхреакцийкинетические и термодинамические показатели, в частностиконстантыдиссоциациипромежуточных фермент-субстратных комплексов,константы скоростииравновесияих образования.
В образовании фермент-субстратных комплексов участвуют водородные связи, электростатические игидрофобные взаимодействия, а в ряде случаев также ковалентные,координационные связи(рис. 4.9). Информация о природе связей междусубстратоми связывающим участкомактивного центра ферментаможет быть получена методамиЭПРиЯМР, а также методами УФ- и ИК-спектроскопии.
Для каталитической активности ферментасущественное значение имеет пространственная структура, в которой жесткие участки α-спиралей чередуются с гибкими, эластичными линейными отрезками, обеспечивающими динамические изменения белковоймолекулыфермента. Этим изме-неням придается большое значение в некоторых теорияхферментативного катализа. Так, в противоположность модели Э. Фишера «ключ-замок» Д. Кошлендом была разработана теория «индуцированного соответствия», допускающая высокую конформационную лабильностьмолекулыбелка-фермента и гибкость и подвижностьактивного центра. Эта теория была основана на весьма убедительных экспериментах, свидетельствующих о том, чтосубстратиндуцирует конформационные изменениямолекулыферментатаким образом, чтоактивный центрпринимает необходимую для связываниясубстратапространственную ориентацию. Иными словами,ферменттолько в присутствии (точнее, в момент присоединения)субстратабудет находиться в активной (напряженной) Т-форме в отличие от неактивной R-формы (рис. 4.10). На рис. 4.10 видно, что присоединениесубстратаS кферментуЕ, вызывая соответствующие измененияконформацииактивного центра, в одних случаях приводит к образованиюактивного комплекса, в других – неактивного комплекса вследствие нарушения пространственного расположенияфункциональных группактивного центрав промежуточном комплексе. Получены экспериментальные доказательства нового положения о том, что постулированное Д. Кошлендом «индуцированное соответствие»субстратаиферментасоздается не обязательно изменениями
43) Биосинтез жирных кислот катализируется синтазой жирных кислот. Эта ферментная система локализована вцитоплазме и нуждается в качестве затравки в ацетил-КоА. В циклической реакции одна молекула удлиняется семикратно на С2-звена. В качестве конечного продукта реакции образуется анион С16-кислоты, пальмитат. Фактический субстрат реакции удлинения цепи малонил-КоА на каждой стадии конденсации отщепляеткарбоксильную группу в вида СО2. Восстановителем в синтезе жирных кислот является НАДФН + Н+. В результате на синтез одной молекулы пальмитата расходуется одна молекула ацетил-КоА, 7 молекулмалонил-КоА и 14 молекул НАДФН + Н+; при этом образуются 7 молекул СО2, 6 молекул H2O, 8 молекул КоА и 14 молекул НАДФ+.
А. Синтаза жирных кислот
Синтаза жирных кислот позвоночных состоит из двух идентичных пептидных цепей, т. е представляет собой гомодимер. Каждая из двух пептидных цепей, представленных на рисунке в виде половинок шара, может катализировать семь различных реакций ([1]-[7]), из которых складывается синтез пальмитата. Пространственное объединение нескольких последовательных реакций в таком мультиферментном комплексе имеет ряд принципиальных преимуществ по сравнению с отдельными ферментами; предотвращаются конкурентные реакции, последовательные реакции согласованы как на конвейере, реакциипротекают особенно эффективно благодаря высокой концентрации субстрата из-за незначительных потерь за счет диффузии.
Каждая половинка синтазы жирных кислот может связывать субстрат тиолсложноэфирной связью (ацильный или ацетильный остаток) по двум SH-группам: цистеинового остатка (Cys-SH) и 4'-фосфопантетеиновой группы (Pan-SH). Pan-SH, очень похожий на кофермент А (см. рис. 111), связан с доменом синтазы, который называют ацилпереносящим белком [АПБ (ACP). Эта часть фермента функционирует как "длинная рука", которая фиксирует субстрат и передает его от одного реакционного центра к другому. Интересно отметить, что реакция при этом зависит от согласованности действия обеих половинок синтазы. Поэтому ферментфункционально активен только в виде димера.
Активность мультиферментного комплекса пространственно распределена по трем различным доменам.Домен 1 катализирует перенос субстратов ацетил-КоА и малонил-КоА [АПБ]-S-ацетилтрансферазой [1] и [АПБ]-S-малонилтрансферазой [2] и последующую конденсацию обоих партнеров 3-оксоацил-[АПБ]-синтазой [3], домен 2 восстанавливает растущую цепь жирной кислоты с помощью 3-оксоацил-[АПБ]-редуктазы [4], 3-гидроксиацил-[АПБ]-дегидратазы [5] и еноил-[АПБ]-редуктазы [6]. Наконец, домен 3 после семи циклов удлинения цепи катализирует высвобождение готового продукта с помощью ацил-[АПБ]-гидролазы [7].
Б. Реакции синтазы жирных кислот
Биосинтез пальмитата (на схеме внизу) начинается с переноса ацетильной группы на уже упомянутый остаток цистеина (Cys-SH) [1] и малонильной группы на 4-фосфопантетеин (Pan-SH) в АПБ [2]. Удлинение цепи происходит вследствие переноса ацетильной группы на углеродный атом С-2 малонильного остатка (голубая стрелка), причем свободная карбоксильная группа отщепляется в виде СО2 [3]. Следующие три стадии реакции, а именно восстановление 3-оксогруппы [4], отщепление воды [5] и вновь восстановление [6], приводят к жирной кислоте с четырьмя углеродными атомами. Ацилтрансфераза [1] переносит этот промежуточный продукт на цистеиновый остаток, освобождая Pan-SH для присоединения следующего малонильного остатка. После семи циклов ацил-[АПБ]-гидролаза [7] «опознает» и освобождает конечный продукт — молекулу пальмитиновой кислоты.
44)гормоны щитовидной железы. Щитовидная железа играет исключительно важную роль в обмене веществ. Об этом свидетельствуют резкое изменение основного обмена, наблюдаемое при нарушениях деятельности щитовидной железы, а также ряд косвенных данных, в частности обильное ее кровоснабжение несмотря на небольшую массу (20–30 г). Щитовидная железа состоит из множества особых полостей – фолликулов, заполненных вязким секретом – коллоидом. В составколлоида входит особый йодсодержащий гликопротеин с высокой мол. массой – порядка 650000 (5000 аминокислотных остатков). Этот глико-протеин получил название йодтиреоглобулина. Он представляет собой запасную форму тироксина и трийодтиронина – основных гормонов фолликулярной части щитовидной железы.
Помимо этих гормонов (биосинтез и функции которых будут рассмотрены ниже), в особых клетках – так называемых парафолликулярных клетках, или С-клетках щитовидной железы, синтезируется гормон пептидной природы, обеспечивающий постоянную концентрацию кальция в крови. Он получил название «кальцитонин». Впервые на существование кальцито-нина, обладающего способностью поддерживать постоянный уровенькальция в крови, указал в 1962 г. Д. Копп, который ошибочно считал, что этот гормон синтезируется паращитовидными железами. В настоящее время кальцитонин не только выделен в чистом виде изткани щитовидной железы животных и человека, но и полностью раскрыта 32-членная аминокислотная последовательность, подтвержденная химическим синтезом. Ниже приведена первичная структура кальцитонина, полученного из щитовидной железы человека: