Файл: Занятие 11 раздел тема патология иммунной системы. Цель занятия.doc
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 25.10.2023
Просмотров: 84
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
Межклеточные взаимодействия при развитии ГЗТ
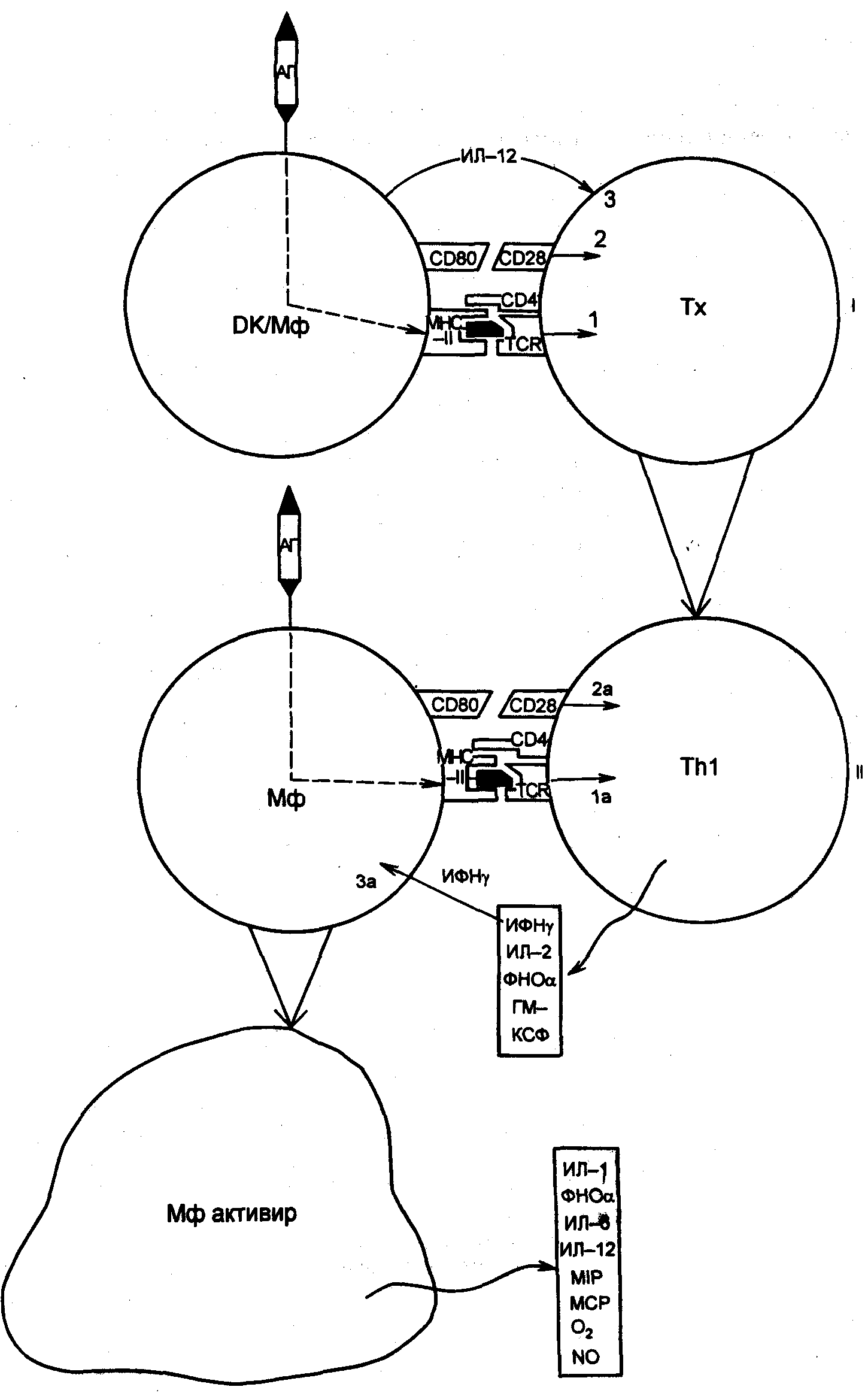
I период: сенсибилизации: АПК ( дендритные клетки- ДК или макрофаги МФ0 взаимодействуют с «наивными» Т- хелперами (Тh0), которые получают 3 сигнала: 1- за счет распознавания с помощью TCR при участии корецептора CD4 комплекса молекулы МНС ІІ класса с антигенным пептидом, образовавшимся в результате молекулы антигена (АГ); 2- за счет взаимодействия костимулирующих молекул CD28 и CD 80/86; 3-за счет действия ИЛ-12, продуцируемого АПК. В результате Т- хелпер дифференцируется в Тh1-клетку.
I I период: разрешения: при повторном попадании АГ в организм.
1 стадия – иммунологическая: в роли АПК выступают МФ. Происходит взаимодействие между МФ, презентирующим пептидный фрагмент того же АГ (сигнал 1а) и зрелым Тh1. При дополнительной костимуляции через CD28 и CD 80/86 (сигнал 2а), клетка активируется и выделяет комплекс цитокинов. Один из них- ИНФγ – активирует макрофаги (сигнал 3а), что приводит к повышению их фагоцитарной, бактерицидной, секреторной и киллерной активности.
2 стадия- патохимическая: активированный Тh1 вырабатывает цитокины :
- γ- ИНФ (главный патохимический медиатор)
- М- КСФ
- ГМ- КСФ
- ИЛ-2 (фактор роста Т- клеток)
- ФХМ (фактор хемотаксиса макрофагов)
- МИФ
3 стадия- патофизиологическая (эффекторная):связана с деятельностью макрофагов. Они выступают как киллерные клетки и вырабатывают:
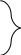
- ИЛ-1
- ФНОα
- АФК гибель патогена
- лизосомальные ферменты
- NO
«Коктейль» цитокинов, выделяемых Тh1-клетками, макрофагами и клетками эндотелия, обусловливает тот широкий спектр общих и местных проявлений, который свойствен ГЗТ. ИЛ-2 обеспечивает размножение Т-клеток. Под влиянием ГМ-КСФ стимулируется миелопоэз, а вырабатываемые макрофагами и эндотелиальными клетками β-хемокины привлекают моноциты в очаг ГЗТ. Характерно, что при этой форме ответа слабо выражена сосудистая реакция и почти отсутствует отек. В то же время происходит сильная инфильтрация очага воспаления клетками, что проявляется в отвердении воспаленного участка.
При развитии неэффективной реакции на внедрение инфекционных агентов, паразитов или инертных частиц, которые не поддаются разрушению и элиминации, формируется гранулема. Гранулема представляет собой морфологическую структуру, в центре которой сосредоточены возбудитель, инфицированные макрофаги, гигантские эпителиоидные клетки (слившиеся макрофаги), детрит, а вокруг формируется клеточный вал, сформированный Т-лимфоцитами или клетками других типов (их состав зависит от природы возбудителя). В центральной части гранулемы обычно формируется некроз, причем распад ткани может вызвать серьезную деструкцию, как это происходит, например, при туберкулезе. Фактически гранулема представляет собой новообразованную морфологическую структуру, предназначенную для изоляции возбудителя или иного чужеродного объекта, разрушение и элиминация которых оказываются невозможными..
Особенности ГЗТ:
• в качестве АПК выступают при ГЗТ при первом действии — дендритные клетки или макрофаги, при повторном — макрофаги.
• Т-хелперами служат при ГЗТ — Тh1-клетки;
• эффекторными клетками при ГЗТ являются— макрофаги, активированные цитокинами Т h1 -клеток.
• период десенсибилизации при ГЗТ
АУТОИММУННАЯ ПАТОЛОГИЯ
Аутоиммунная патология развивается тогда, когда иммунная система реагирует на собственные, неизмененные антигены. Т.О. – это болезнь иммунной системы, поскольку агрессия развивается против неизмененных АГ. В норме реакция на неизмененные АГ организма со стороны иммунной системы отсутствует. Это связано с существованием иммунологической толерантности.
Иммунологическая толерантность приобретается Т- клетками в тимусе, в процессе эмбриогенеза. В основе лежат процессы «+» селекции клона или клональной экспансии и «-» селекции клона или клональной делеции и клональной анергии.
АПОПТОЗ И ФОРМИРОВАНИЕ ВТОРИЧНОГО КЛОНАЛЬНОГО РЕПЕРТУАРА Т-ЛИМФОЦИТОВ
-
Первичный (антигенраспознающий) репертуар - результат событий на стадии СД 4- 8- тимоцитов, реализуется на уровне генов и клеточной мембраны и проявляется экспрессией на поверхности клеток Т-клеточного рецептора (ТСR) с индивидуальной специфичностью. -
Переход незрелых кортикальных тимоцитов в стадию созревания СД 4+ 8+ сопровождается экспрессией на их поверхности Fas-рецептора (Fas-R), что в сочетании с низкой экспрессией Bcl 2 делает их высокочувствительными к индукторам апоптоза, в частности, к Fas L. -
Положительная селекция клонов осуществляется в глубоких слоях коры тимуса при взаимодействии тимоцитов с эпителиальными клетками (ЭК), несущими на поверхности молекулы МНС II класса и экспрессирующими Fas L. Контакт Fas L ЭК и Fas-R тимоцитов включает у последних механизм апоптоза, обрекая их на гибель. Процесс может быть остановлен сигналом из участков взаимодействия ТСR с молекулами МНС ЭК. Однако, сигнал поступит лишь в том случае, если ТСR будет комплементарен к этим молекулам, независимо от того, в сочетании с каким (аутологичным и гетерологичным пептидом) они способны его распознавать. Т.о. к дальнейшему развитию будут допущены лишь те клоны тимоцитов, которые обладают той или иной степенью сродства к продуктам аутологичных генов МНС. -
Отрицательная селекция осуществляется в мозговом слове и кортико-медуллярной зоне тимуса в процессе взаимодействия тимоцитов с дендритными клетками (ДК), несущими МНС I и II класса. Тимоциты по-прежнему экспрессируют Fas-R, но способны к (слабой) продукции Bcl 2. Поэтому их контакт с ДК, несущими Fas L недостаточен для развития апоптоза. Требуется костимулирующий сигнал. Этот сигнал поступает из участка связывания ТСR с аутологичным антигенным комплексом, состоящим из аутологичного пептида на "своей" молекуле МНС. Такая комплементарность инициирует активацию незрелых тимоцитов, что необходимо для реализации апоптоза ("активационный апоптоз"). В итоге выживают лишь те клоны лимфоцитов, которые распознают комплексы из чужеродных антигенов с аутологичными молекулами МНС. Завершается формирование вторичного клонального репертуара Т-лимфоцитов.
Иммунологическая толерантность формируется не ко всем АГ организма. Существуют т.н. забарьерные АГ, к которым нет толерантности. К забарьерным АГ относятся:
- хрусталик
- сперматозоиды
- коллоид щитовидной железы
- миелин нервных волокон
До тех пор, пока существует физиологический барьер и нет контакта между АГ и лимфоцитами, аутоиммунная патология не развивается.
Н
 ельзя сказать, что в организме полностью отсутствуют аутореактивные клетки. Они сохраняются, чтобы устранять продукты тканевой деструкции – это аутоиммунные реакции. Выраженность их проявлений лимитируется супресорными механизмами (Тh3 ИЛ-10).
ельзя сказать, что в организме полностью отсутствуют аутореактивные клетки. Они сохраняются, чтобы устранять продукты тканевой деструкции – это аутоиммунные реакции. Выраженность их проявлений лимитируется супресорными механизмами (Тh3 ИЛ-10).Если организму наносится вред аутоагрессией, то это аутоиммунные заболевания (АИЗ).
МЕХАНИЗМЫ РАЗВИТИЯ АИЗ



Недостаточно сформированная
толерантность
(дефект FAS- зависимого апоптоза) Сформированная
Не сформированная толерантность толерантность
 (забарьерные АГ)
(забарьерные АГ) Срыв иммунологической толерантности
Срыв иммунологической толерантностиНарушение барьеров Механизмы срыва иммунологической толерантности
-
А нтигенная мимикрия -
Поликлональная митогенная активация -
Аномальная экспрессия МНСII на неиммуннокомпетентных клетках
Аутоагрессия - Точковые мутации в кодоне клеток
-
Дефицит Т- супрессорных влияний
В основе аутоиммунных процессов могут лежат клеточные или гуморальные иммунные механизмы, что определяется (как и в случае иммунной защиты) преобладающей пусковой ролью Т-хелперов типов Th1 или Th2. Thl-зависимые аутоиммунные процессы регистрируются особенно часто. Они имеют два основных варианта в зависимости от того, какой тип эффекторных клеток при них активируется — С08+-киллеры или С04
+-клетки — продуценты цитокинов. В первом случае включается цитотоксический механизм поражения (например, при инсулинзависимом диабете), связанный с активностью Т-киллеров, во втором — основой патологического процесса является реакция типа ГЗТ (при рассеянном склерозе, ревматоидном артрите), которая осуществляется при участии Т-хелперов и макрофагов. Цитотоксический механизм обусловливает более локализованный тип поражения, например при инсулинзависимом диабете. Напротив, процессы, сопряженные с развитием ГЗТ, вовлекают более значительные массивы тканей (например, при ревматоидном артрите).
Реже встречаются Тп2-зависимые аутоиммунные процессы. Ведущая патогенетическая роль аутоантител показана при системной красной волчанке, аутоиммунной гемолитической анемии, идиопатической тромбоцитопенической пурпуре и ряде других цитопений, тяжелой миастении и др. Для аутоиммунных процессов гуморального типа характерно накопление аутоантител преимущественно lgG-класса, которые обладают способностью вовлекать в процесс другие факторы — гуморальные (комплемент) и клеточные (макрофаги, NK-клетки). Поэтому патогенный эффект аутоантител обычно реализуется с участием цитотоксического механизма (при гемолитической анемии и других аутоиммунных поражениях клеток крови). Реже проявляется стимулирующий эффект аутоантител; при токсическом зобе (Базедова болезнь) и возможно присоединение элементов тироидита.
Возможно также развитие аутоиммунной патологии по иммуноком-плексному типу, например при системной волчанке. Аутоантигенами в этом случае служат широко распространенные молекулы — нативная ДНК, белки межклеточного вещества (особенно коллаген) и т.д., которые взаимодействуют с аутоантителами и формируют иммунные комплексы in situ (фиксированные в тканях). Взаимодействие с иммунными комплексами активирует клетки иммунной системы (макрофаги, NK-клетки и т.д.), что обусловливает развитие локального воспаления и проявление цитотоксичности.
Таким образом, основой аутоиммунных процессов служит развитие иммунного ответа на собственные (аутологичные) антигены организма. Эти процессы возникают при нарушении (часто наследственно обусловленном) центральных или периферических механизмов, обеспечивающих запрет на реакции иммунной системы против аутологичных антигенов. В зависимости от локализации аутоантигенов различают органоспецифи-ческие и системные аутоиммунные процессы, а в зависимости от преобладающих механизмов — клеточно-опосредованные и гуморальные.
ИММУНОДЕФИЦИТНЫЕ СОСТОЯНИЯ
Первичные иммунодефициты
Группу первичных иммунодефицитов образуют заболевания, в основе которых лежит наследственно обусловленная дефектность структуры и функционирования иммунной системы, которая проявляется в нарушении иммунной защиты.
Первичные иммунодефициты — это очень редкие состояния (примерно 1 больной на 1 000 000 человек). Они являются почти исключительно уделом детского возраста, поскольку значительная часть больных с тяжелыми формами иммунодефицитов не доживает до 20 лет, а при более легких формах иммунологические дефекты с возрастом в определенной степени компенсируются.
Как правило, в основе первичных иммунодефицитов лежит генетически обусловленный блок развития клеток иммунной системы или выпадение важных иммунных процессов вследствие дефекта определенных молекул, например ферментов или мембранных структур (схема 1).
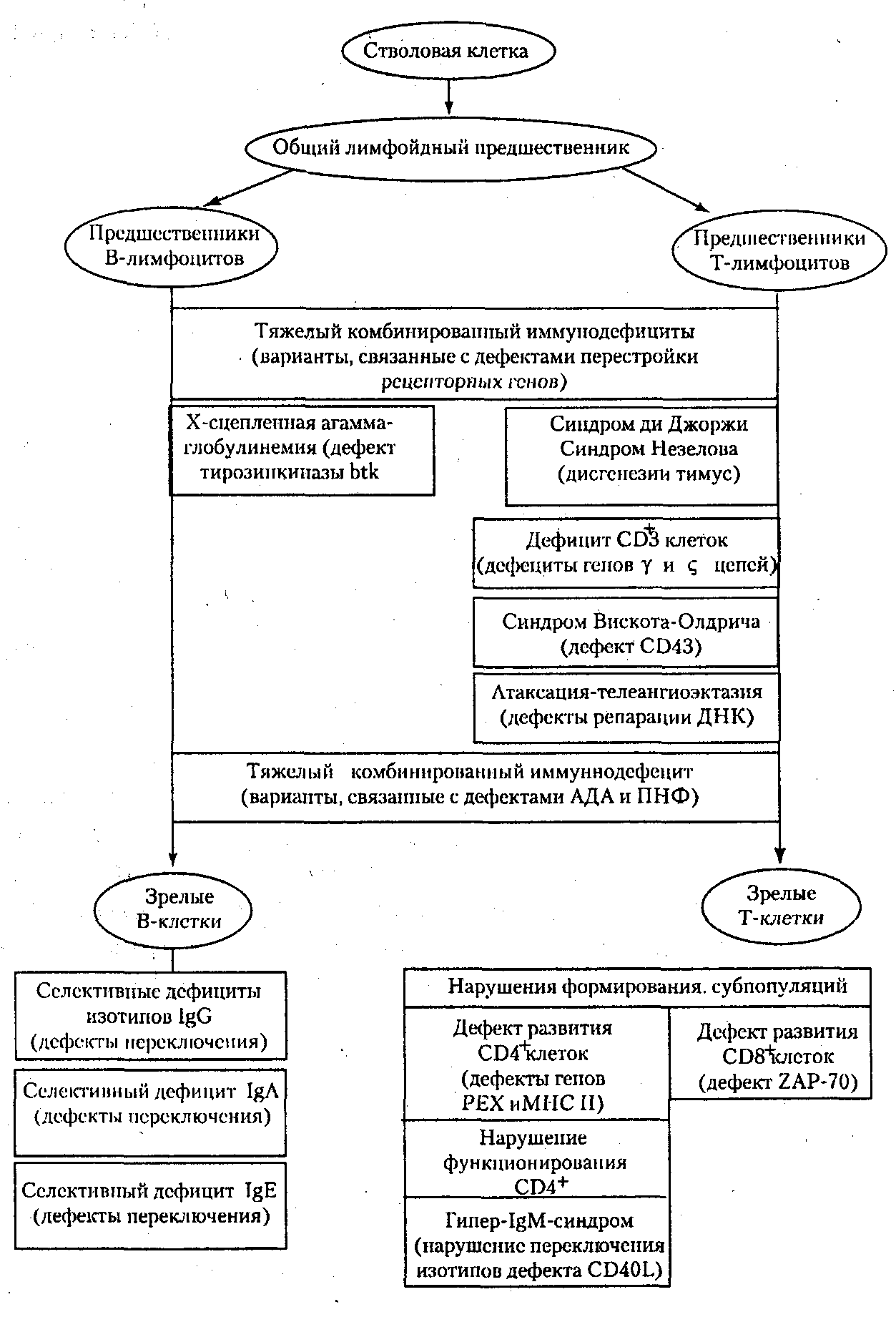
Первичные иммунодефициты можно разделить по преобладающему типу поражений звеньев иммунной системы на 3 типа:
— комбинированные иммунодефициты;
— иммунодефициты с преимущественным поражением клеточного иммунитета;
— преимущественно гуморальные иммунодефициты.
К первым относят заболевания, в основе которых лежат генетические дефекты, затрагивающие различные линии дифференцировки лимфоцитов, а также ранние этапы их развития, общие для Т- и В-линий. Во вторую группу входят иммунодефициты, при которых нарушается развитие Т-клеток и страдают опосредуемые ими реакции клеточного иммунитета; к этой же группе относятся дефекты фагоцитирующих клеток. В группу гуморальных иммунодефицитов включают патологию, в основе которой лежит нарушение развития В-клеток и Т-хелперов гуморального ответа, а также патологию компонентов комплемента.
В последние годы выясняются молекулярные основы поражения при первичных иммунодефицитах. Одной из первых была расшифрована природа комбинированных иммунодефицитов, связанных с недостаточностью ферментов пуринового метаболизма. Известны варианты таких дефектов, обусловленные мутациями генов, кодирующих аденозиндезаминазу и пуриннуклеотидфосфорилазу. Основой другой формы тяжелого комбинированного иммунодефицита, затрагивающего Т- и В-ростки лимфопоэза, служит дефект процесса перестройки генов антигенраспознающих рецепторов,