ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 04.02.2024
Просмотров: 1654
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
СОДЕРЖАНИЕ
ЖИРОВЫЕ СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ
УГЛЕВОДНЫЕ СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ
НАРУШЕНИЯ ОБМЕНА ПРОТЕИНОГЕННЫХ ПИГМЕНТОВ
НАРУШЕНИЯ ОБМЕНА ЛИПИДОГЕННЫХ ПИГМЕНТОВ
НАРУШЕНИЯ ОБМЕНА НУКЛЕОПРОТЕИНОВ
Различия злокачественных и доброкачественных новообразований
Стадии заболевания лимфогранулематозом
ЦЕРЕБРОВАСКУЛЯРНЫЕ ЗАБОЛЕВАНИЯ
Постнекротический цирроз печени (крупноузловой, мультибульбарный)
Классификация гломерулонефрита
Что такое Острый диффузный гломерулонефрит -
Патогенез (что происходит?) во время Острого диффузного гломерулонефрита:
Диагностика
Для дифференциальной диагностики хронического лимфоцитарного лейкоза с другими лимфопролиферативными заболеваниями необходимо проанализировать количество В-клеток в периферической крови, мазок крови и провести иммунофенотипирование циркулирующих в крови лимфоцитов. Дополнительно для определения прогноза (но не схемы лечения) иногда проводят цитогенетическое исследование, определяют мутационный статус локуса IgVH, количество ZAP-70 или CD38 в клетках ХЛЛ, количество CD23, тимидинкиназы и β2-микроглобулина в сыворотке крови, а также анализируют биоптат или аспират костного мозга[12].
Анализ крови
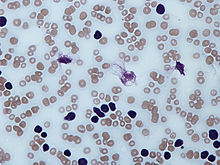
Тени Гумпрехта в мазке крови
Необходимым критерием диагноза хронического лимфоцитарного лейкоза является повышение абсолютного числа В-лимфоцитов в крови до или более 5×109/л. Кроме того, эти лимфоциты должны иметь характерный иммунофенотип: на их поверхности должны обнаруживаться CD19, CD5, CD23, небольшие количества CD20 и CD79b, а также лёгкие цепи иммуноглобулинов[13].
В мазке крови обнаруживаются опухолевые клетки, которые имеют морфологию зрелых (малых) лимфоцитов: «штампованное» ядро с конденсированным хроматином без ядрышка, узкий ободок цитоплазмы. Характерно наличие так называемых теней Гумпрехта, которые представляют собой лейкозные клетки, разрушившиеся в процессе приготовления мазка. Помимо малых лимфоцитов в мазке могут присутствовать более крупные или атипичные клетки, иногда отмечается существенная (более 10 %) примесь омоложенных клеток (пролимфоцитов и параиммунобластов), требующая проведения дифференциального диагноза с пролимфоцитарным лейкозом[12].
Иммунофенотипирование
Иммунофенотипирование лимфоцитов методом проточной цитометрии обязательно для подтверждения диагноза. Высокочувствительная проточная цитометрия позволяет обнаруживать одну злокачественную клетку на 10 000 нормальных лейкоцитов[13]. В качестве диагностического материала обычно используется периферическая кровь. Для клеток ХЛЛ характерен аберрантный иммунофенотип: одновременная экспрессия (коэкспрессия) Т-клеточного маркера CD5 и В-клеточных маркеров CD19 и CD23[12]. Количество В-клеточных маркеров CD20, CD79b и мембраносвязанных иммуноглобулинов IgM и IgD понижено по сравнению с нормальными В-клетками[1]. В дополнение к этому выявляется клональность. Диагноз ХЛЛ также может быть установлен на основании данных иммуногистохимического исследования биоптата лимфатического узла или селезёнки.
Подозрение на хронический лимфоцитарный лейкоз также возникает в случае обнаружения у в остальном здоровых людей увеличения абсолютного числа клональных B-лимфоцитов соответствующего иммунофенотипа, даже если общее их количество в периферической крови меньше 5000/микролитр. Если этому признаку не сопутствует лимфаденопатия или органомегалия, цитопении или другие признаки заболевания, такое состояние диагностируется как моноклональный B-лимфоцитоз[12]. Согласно исследованию, проведённому на 1520 участниках в возрасте от 62 до 80 лет с нормальными показателями крови, моноклональный B-лимфоцитоз с иммунофенотипом ХЛЛ обнаруживается у 5 % людей в этой возрастной группе. Такой лимфоцитоз может прогрессировать в ХЛЛ со скоростью около 1 % в год[13].
Цитогенетическое исследование
Цитогенетическое исследование проводится методом стандартного кариотипирования или FISH. Задача исследования — выявление хромосомных мутаций, часть из которых имеет прогностическую значимость. Из-за возможности клональной эволюции исследование должно повторяться перед каждой линией терапии и в случае возникновения рефрактерности.
Стандартное кариотипирование возможно только для клеток в метафазе клеточного цикла. Так как злокачественные клетки при ХЛЛ обладают низкой митотической активностью, для получения необходимого для анализа количества метафаз требуется применение митогенов. Но даже в таком случае хромосомные мутации удаётся обнаружить только в 40—50 % случаев[14].
Интерфазная FISH при хроническом лимфоцитарном лейкозе не требует применения митогенов и отличается большей чувствительностью. При анализе используют локус-специфичные зонды, позволяющие выявлять наиболее распространённые хромосомные перестройки (как правило делеции). Этот метод позволяет детектировать хромосомные мутации в более чем 80 % случаев хронического лимфоцитарного лейкоза[14].
У каждого отдельного пациента может быть обнаружена одна, две и более стандартных мутации. Исследование, проведённое на 325 пациентах с хроническим лимфоцитарным лейкозом, позволило установить иерархию кариопитов по их прогностической способности: del17p, del11q, трисомия 12, нормальный кариотип и del13q. Если у пациента обнаружено больше одной мутации, то прогноз делают по той из них, которая находится выше в этом списке[14].
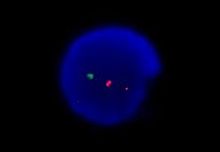
Результат FISH-исследования. Определяется только один аллель гена ATM (зеленая метка). У пациента имеется del11q22.3.
Хромосомные перестройки ассоциированы с определёнными клиническими характеристиками заболевания[14]:
-
del13q14 выявляется в
55 % случаев, делеция может быть моно- и биаллельной, заболевание, как правило, диагностируется на ранней стадии и развивается медленно, прогноз благоприятный;
Диагностика
Для дифференциальной диагностики хронического лимфоцитарного лейкоза с другими лимфопролиферативными заболеваниями необходимо проанализировать количество В-клеток в периферической крови, мазок крови и провести иммунофенотипирование циркулирующих в крови лимфоцитов. Дополнительно для определения прогноза (но не схемы лечения) иногда проводят цитогенетическое исследование, определяют мутационный статус локуса IgVH, количество ZAP-70 или CD38 в клетках ХЛЛ, количество CD23, тимидинкиназы и β2-микроглобулина в сыворотке крови, а также анализируют биоптат или аспират костного мозга[12].
Анализ крови
Тени Гумпрехта в мазке крови
Необходимым критерием диагноза хронического лимфоцитарного лейкоза является повышение абсолютного числа В-лимфоцитов в крови до или более 5×109/л. Кроме того, эти лимфоциты должны иметь характерный иммунофенотип: на их поверхности должны обнаруживаться CD19, CD5, CD23, небольшие количества CD20 и CD79b, а также лёгкие цепи иммуноглобулинов[13].
В мазке крови обнаруживаются опухолевые клетки, которые имеют морфологию зрелых (малых) лимфоцитов: «штампованное» ядро с конденсированным хроматином без ядрышка, узкий ободок цитоплазмы. Характерно наличие так называемых теней Гумпрехта, которые представляют собой лейкозные клетки, разрушившиеся в процессе приготовления мазка. Помимо малых лимфоцитов в мазке могут присутствовать более крупные или атипичные клетки, иногда отмечается существенная (более 10 %) примесь омоложенных клеток (пролимфоцитов и параиммунобластов), требующая проведения дифференциального диагноза с пролимфоцитарным лейкозом[12].
Иммунофенотипирование
Иммунофенотипирование лимфоцитов методом проточной цитометрии обязательно для подтверждения диагноза. Высокочувствительная проточная цитометрия позволяет обнаруживать одну злокачественную клетку на 10 000 нормальных лейкоцитов[13]. В качестве диагностического материала обычно используется периферическая кровь. Для клеток ХЛЛ характерен аберрантный иммунофенотип: одновременная экспрессия (коэкспрессия) Т-клеточного маркера CD5 и В-клеточных маркеров CD19 и CD23[12]. Количество В-клеточных маркеров CD20, CD79b и мембраносвязанных иммуноглобулинов IgM и IgD понижено по сравнению с нормальными В-клетками[1]. В дополнение к этому выявляется клональность. Диагноз ХЛЛ также может быть установлен на основании данных иммуногистохимического исследования биоптата лимфатического узла или селезёнки.
Подозрение на хронический лимфоцитарный лейкоз также возникает в случае обнаружения у в остальном здоровых людей увеличения абсолютного числа клональных B-лимфоцитов соответствующего иммунофенотипа, даже если общее их количество в периферической крови меньше 5000/микролитр. Если этому признаку не сопутствует лимфаденопатия или органомегалия, цитопении или другие признаки заболевания, такое состояние диагностируется как моноклональный B-лимфоцитоз[12]. Согласно исследованию, проведённому на 1520 участниках в возрасте от 62 до 80 лет с нормальными показателями крови, моноклональный B-лимфоцитоз с иммунофенотипом ХЛЛ обнаруживается у 5 % людей в этой возрастной группе. Такой лимфоцитоз может прогрессировать в ХЛЛ со скоростью около 1 % в год[13].
Цитогенетическое исследование
Цитогенетическое исследование проводится методом стандартного кариотипирования или FISH. Задача исследования — выявление хромосомных мутаций, часть из которых имеет прогностическую значимость. Из-за возможности клональной эволюции исследование должно повторяться перед каждой линией терапии и в случае возникновения рефрактерности.
Стандартное кариотипирование возможно только для клеток в метафазе клеточного цикла. Так как злокачественные клетки при ХЛЛ обладают низкой митотической активностью, для получения необходимого для анализа количества метафаз требуется применение митогенов. Но даже в таком случае хромосомные мутации удаётся обнаружить только в 40—50 % случаев[14].
Интерфазная FISH при хроническом лимфоцитарном лейкозе не требует применения митогенов и отличается большей чувствительностью. При анализе используют локус-специфичные зонды, позволяющие выявлять наиболее распространённые хромосомные перестройки (как правило делеции). Этот метод позволяет детектировать хромосомные мутации в более чем 80 % случаев хронического лимфоцитарного лейкоза[14].
У каждого отдельного пациента может быть обнаружена одна, две и более стандартных мутации. Исследование, проведённое на 325 пациентах с хроническим лимфоцитарным лейкозом, позволило установить иерархию кариопитов по их прогностической способности: del17p, del11q, трисомия 12, нормальный кариотип и del13q. Если у пациента обнаружено больше одной мутации, то прогноз делают по той из них, которая находится выше в этом списке[14].
Результат FISH-исследования. Определяется только один аллель гена ATM (зеленая метка). У пациента имеется del11q22.3.
Хромосомные перестройки ассоциированы с определёнными клиническими характеристиками заболевания[14]:
-
del13q14 выявляется в
55 % случаев, делеция может быть моно- и биаллельной, заболевание, как правило, диагностируется на ранней стадии и развивается медленно, прогноз благоприятный;
Диагностика
Для дифференциальной диагностики хронического лимфоцитарного лейкоза с другими лимфопролиферативными заболеваниями необходимо проанализировать количество В-клеток в периферической крови, мазок крови и провести иммунофенотипирование циркулирующих в крови лимфоцитов. Дополнительно для определения прогноза (но не схемы лечения) иногда проводят цитогенетическое исследование, определяют мутационный статус локуса IgVH, количество ZAP-70 или CD38 в клетках ХЛЛ, количество CD23, тимидинкиназы и β2-микроглобулина в сыворотке крови, а также анализируют биоптат или аспират костного мозга[12].
Анализ крови
Тени Гумпрехта в мазке крови
Необходимым критерием диагноза хронического лимфоцитарного лейкоза является повышение абсолютного числа В-лимфоцитов в крови до или более 5×109/л. Кроме того, эти лимфоциты должны иметь характерный иммунофенотип: на их поверхности должны обнаруживаться CD19, CD5, CD23, небольшие количества CD20 и CD79b, а также лёгкие цепи иммуноглобулинов[13].
В мазке крови обнаруживаются опухолевые клетки, которые имеют морфологию зрелых (малых) лимфоцитов: «штампованное» ядро с конденсированным хроматином без ядрышка, узкий ободок цитоплазмы. Характерно наличие так называемых теней Гумпрехта, которые представляют собой лейкозные клетки, разрушившиеся в процессе приготовления мазка. Помимо малых лимфоцитов в мазке могут присутствовать более крупные или атипичные клетки, иногда отмечается существенная (более 10 %) примесь омоложенных клеток (пролимфоцитов и параиммунобластов), требующая проведения дифференциального диагноза с пролимфоцитарным лейкозом[12].
Иммунофенотипирование
Иммунофенотипирование лимфоцитов методом проточной цитометрии обязательно для подтверждения диагноза. Высокочувствительная проточная цитометрия позволяет обнаруживать одну злокачественную клетку на 10 000 нормальных лейкоцитов[13]. В качестве диагностического материала обычно используется периферическая кровь. Для клеток ХЛЛ характерен аберрантный иммунофенотип: одновременная экспрессия (коэкспрессия) Т-клеточного маркера CD5 и В-клеточных маркеров CD19 и CD23[12]. Количество В-клеточных маркеров CD20, CD79b и мембраносвязанных иммуноглобулинов IgM и IgD понижено по сравнению с нормальными В-клетками[1]. В дополнение к этому выявляется клональность. Диагноз ХЛЛ также может быть установлен на основании данных иммуногистохимического исследования биоптата лимфатического узла или селезёнки.
Подозрение на хронический лимфоцитарный лейкоз также возникает в случае обнаружения у в остальном здоровых людей увеличения абсолютного числа клональных B-лимфоцитов соответствующего иммунофенотипа, даже если общее их количество в периферической крови меньше 5000/микролитр. Если этому признаку не сопутствует лимфаденопатия или органомегалия, цитопении или другие признаки заболевания, такое состояние диагностируется как моноклональный B-лимфоцитоз[12]. Согласно исследованию, проведённому на 1520 участниках в возрасте от 62 до 80 лет с нормальными показателями крови, моноклональный B-лимфоцитоз с иммунофенотипом ХЛЛ обнаруживается у 5 % людей в этой возрастной группе. Такой лимфоцитоз может прогрессировать в ХЛЛ со скоростью около 1 % в год[13].
Цитогенетическое исследование
Цитогенетическое исследование проводится методом стандартного кариотипирования или FISH. Задача исследования — выявление хромосомных мутаций, часть из которых имеет прогностическую значимость. Из-за возможности клональной эволюции исследование должно повторяться перед каждой линией терапии и в случае возникновения рефрактерности.
Стандартное кариотипирование возможно только для клеток в метафазе клеточного цикла. Так как злокачественные клетки при ХЛЛ обладают низкой митотической активностью, для получения необходимого для анализа количества метафаз требуется применение митогенов. Но даже в таком случае хромосомные мутации удаётся обнаружить только в 40—50 % случаев[14].
Интерфазная FISH при хроническом лимфоцитарном лейкозе не требует применения митогенов и отличается большей чувствительностью. При анализе используют локус-специфичные зонды, позволяющие выявлять наиболее распространённые хромосомные перестройки (как правило делеции). Этот метод позволяет детектировать хромосомные мутации в более чем 80 % случаев хронического лимфоцитарного лейкоза[14].
У каждого отдельного пациента может быть обнаружена одна, две и более стандартных мутации. Исследование, проведённое на 325 пациентах с хроническим лимфоцитарным лейкозом, позволило установить иерархию кариопитов по их прогностической способности: del17p, del11q, трисомия 12, нормальный кариотип и del13q. Если у пациента обнаружено больше одной мутации, то прогноз делают по той из них, которая находится выше в этом списке[14].
Результат FISH-исследования. Определяется только один аллель гена ATM (зеленая метка). У пациента имеется del11q22.3.
Хромосомные перестройки ассоциированы с определёнными клиническими характеристиками заболевания[14]:
-
del13q14 выявляется в
трисомия по хромосоме 12 выявляется в 15 % случаев, прогноз обычный;
del11q выявляется в 15 % случаев, болезнь диагностируют на более поздних стадиях, выше вероятность проявления конституциональных симптомов, болезнь быстро прогрессирует, прогноз неблагоприятный, данная мутация может ассоциироваться с резистентностью к алкилирующим химиопрепаратам;
del17p13 выявляется в 7 % случаев, болезнь диагностируют на более поздних стадиях, выше вероятность проявления конституциональных симптомов, болезнь быстро прогрессирует, прогноз наиболее неблагоприятный, клоны часто бывают устойчивы к стандартным схемам химиотерапии с использованием алкилирующих препаратов и/или аналогов пурина[12];
del6q21 характеризуется неблагоприятным прогнозом[1].
По клиническим проявлениям хронический лимфоцитарный лейкоз является довольно гетерогенным заболеванием: болезнь может протекать длительно без прогрессии или, наоборот, очень агрессивно[5]. Примерно в 30 % случаев болезнь прогрессирует медленно, так что смерть наступает по причине, не связанной с болезнью. В 15 % случаев смерть от болезни и/или побочных эффектов лечения наступает в течение 2—3 лет с момента постановки диагноза. В остальных случаях болезнь медленно прогрессирует в течение 5—10 лет, после чего наступает терминальная стадия заболевания, за которой следует смерть[28]. В случае пациентов из группы низкого риска медиана выживаемости от момента постановки диагноза достигает 8—10 лет. Известен ряд факторов, которые позволяют прогнозировать результаты лечения и продолжительность жизни, в том числе:
-
Наличие или отсутствие признаков соматической гипермутации в генах вариабельных фрагментов иммуноглобулинов В-клеточного рецептора, -
Использование определенных V-генов в структуре В-клеточного рецептора (например, VH3—21), -
Уровень экспрессии тирозинкиназы Zap-70, -
Уровень экспрессии поверхностного маркера CD38, -
Хромосомные мутации del17p, del11q, затрагивающие гены TP53 и ATM, -
Уровень бета-2-микроглобулина в сыворотке крови, -
Стадия заболевания по Rai и Binet, -
Время удвоения числа лимфоцитов периферической крови и т. д.
Опухолевая трансформация, при которой клетки клона приобретают новые характеристики, делающие их похожими на диффузную крупноклеточную лимфому, носит название синдром Рихтера. Прогноз при наличии трансформации крайне неблагоприятный.
115. Охарактеризуйте парапротеинемические лейкозы: разновидности, функционально-морфологические особенности, патологоанатомическая диагностика.
Парапротеинемические лейкозы — группа заболеваний, относящихся к хроническим лимфоцитарным лейкозам. В эту группу входят три заболевания – миеломная болезнь, первичная макроглобулинемия Вальденстрёма и болезнь тяжелых цепей [1]Франклина.
Другое название — злокачественные иммунопролиферативные заболевания.
Особенностью парапротеинемических лейкозов является способность опухолевых клеток синтезировать однородные иммуноглобулины или их фрагменты – парапротеины [2] (это связано с цитогенезом опухолевых клеток). Опухолевые клетки при парапротеинемических лейкозах дифференцируются по плазмоцитарному типу, сохраняя в извращенной форме особенность плазматических клеток синтезировать иммуноглобулины. Особое место среди парапротеинемических лейкозов имеет миеломная болезнь.
В эту группу входят три заболевания: миеломная болезнь (Рустицкого-Каллера), первичная макроглобулинемия (Вальден- стрема), болезнь тяжелых цепей (Франклина). Особенностью па- рапротеинемических лейкозов является способность опухолевых клеток синтезировать патологические белки - парапротеины. Опухолевые клетки при парапротеинемических лейкозах дифференцируются по плазмоцитарному типу, сохраняя в извращенной форме особенность плазматических клеток синтезировать иммуноглобулины. Наибольшее значение среди парапротеинемических лейкозов имеет миеломная болезнь.
МИЕЛОМНАЯ БОЛЕЗНЬ (болезнь Рустицкого-Калера, множественная миелома, генерализованная плазмоцитома) встречается в основном у взрослых. Описаны единичные наблюдения у людей моложе 30 лет. Свое название заболевание и опухолевая клетка получили в связи с преимущественной локализацией процесса на территории костного мозга [миелон - костный мозг). Выделяют несколько вариантов миеломной болезни в зависимости от характера распространения миеломных инфильтратов в костном мозге, от разновидности миеломных клеток и от типа синтезируемого парапротеина.
По характеру распространенности опухолевого инфильтрата в костном мозге выделяют диффузную, диффузно-узловатую, множественно-узловатую формы миеломы. По клеточному составу - плазмоцитарную
, плазмобластную, полиморфноклеточную и мелкоклеточную миелому.
Опухолевая ткань разрастается преимущественно в плоских костях [череп, ребра, таз) и в позвоночнике. В костях возникают остеолизис, остеопороз, пазушное рассасывание, что нередко приводит к патологическим переломам костей.
Белок попадает в кровь и в ней возникает гиперпротеинемия, диспротеинемия, парапротеинемия. Белок выделяется мочой, поэтому очень характерна парапротеинурия. Белок всасывается канальцами почек, поэтому в них может возникать миеломная почка [парапротеинемический нефроз), амилоидоз и пиелонефрит, что часто приводит к уремии
116. Охарактеризуйте регионарные опухолевые заболевания кроветворной и лимфатический ткани (лим-фомы): этиология, патогенез, морфология.
Лимфомы - регионарные опухолевые заболевания кроветворной и лимфатической ткани |
| |
В эту группу заболеваний входят лимфосаркома, грибовидный микоз, болезнь Сезари, ретикулосаркома, лимфогранулематоз (болезнь Ходжкина).
Лимфомы могут быть В-клеточного и Т-клеточного происхождения. На этом основана классификация лимфом, предложенная Люкез и Коллинз. Согласно этой классификации, В-клеточные лимфомы могут быть: мелкоклеточными (В), центроцитарными, иммунобластными (В), плазмолимфоцитарными, а Т-клеточные лимфомы - мелкоклеточными (Т), из лимфоцитов с перекрученными ядрами, иммунобластными (Т), а также представлены грибовидным микозом и болезнью Сезари. Кроме того, выделяют неклассифицируемые лимфомы. Из этой классификации следует, что мелкоклеточные и иммунобластные лимфомы могут происходить как из В-, так и из Т-клеток. Только из В-клеток развиваются центроцитарная и плазмолимфоцитарная лимфомы и только из Т-клеток - лимфома из лимфоцитов с перекрученными ядрами, грибовидный микоз и болезнь Сезари.
Этиология и патогенез. Лимфомы не имеют каких-либо особенностей по сравнению с лейкозами. Следует подчеркнуть, что в условиях современной терапии цитостатическими средствами некоторые лимфомы (лимфосаркома) нередко «завершают» терминальную стадию лейкоза. Вместе с тем сами они способны «трансформироваться» в лейкоз. Из этого следует, что разграничение опухолей системы крови на «диффузные» и «регионарные», необходимое в интересах нозологии, с позиций онкогенеза весьма условно.
Патологическая анатомия. Каждая из лимфом имеет характерную морфологическую картину.
Лимфосаркома - злокачественная опухоль, возникающая из клеток лимфоцитарного ряда. При этой опухоли поражаются лимфатические узлы, причем чаще - медиастинальные и забрюшинные, реже - паховые и подмышечные. Возможно развитие опухоли в лимфатической ткани желудочно-кишечного тракта, селезенке и других органах. Вначале опухоль носит локальный, ограниченный характер. Лимфатические узлы резко увеличиваются, спаиваются между собой и образуют пакеты, которые сдавливают окружающие ткани. Узлы плотные, на разрезе серо-розовые, с участками некроза и кровоизлияний. В дальнейшем происходит генерализация процесса, т. е. лимфогенное и гематогенное метастазирование с образованием множественных отсевов в лимфатических узлах, легких, коже, костях и других органах. В лимфатических узлах разрастаются опухолевые клетки типа В- или Т-лимфоцитов, пролимфоцитов, лимфобластов, иммунобластов.