ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 04.02.2024
Просмотров: 1618
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
СОДЕРЖАНИЕ
ЖИРОВЫЕ СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ
УГЛЕВОДНЫЕ СТРОМАЛЬНО-СОСУДИСТЫЕ ДИСТРОФИИ
НАРУШЕНИЯ ОБМЕНА ПРОТЕИНОГЕННЫХ ПИГМЕНТОВ
НАРУШЕНИЯ ОБМЕНА ЛИПИДОГЕННЫХ ПИГМЕНТОВ
НАРУШЕНИЯ ОБМЕНА НУКЛЕОПРОТЕИНОВ
Различия злокачественных и доброкачественных новообразований
Стадии заболевания лимфогранулематозом
ЦЕРЕБРОВАСКУЛЯРНЫЕ ЗАБОЛЕВАНИЯ
Постнекротический цирроз печени (крупноузловой, мультибульбарный)
Классификация гломерулонефрита
Что такое Острый диффузный гломерулонефрит -
Патогенез (что происходит?) во время Острого диффузного гломерулонефрита:
бородавчатый эндокардит, названный по имени описавших его авторов эндокардитом Либмана и Сакса.
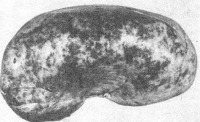
Рис. 179. Почка при волчаночном нефрите.
Как уже говорилось, сосуды разного калибра подвергаются значительным изменениям, особенно сосуды микроциркуляторного русла — возникают артериолиты, капилляриты и венулиты. В стенке аорты в связи с поражением её микрососудов появляются вторичные изменения в виде эластолиза и мелких рубчиков в средней оболочке. В разных органах васкулиты вызывают вторичные изменения — дистрофию паренхиматозных элементов, некроз.
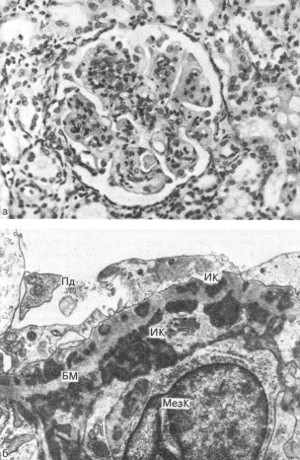
Рис. 180. Волчаночный нефрит. а — кариорексис, гиалиновые тромбы (гистологическая картина); б — субэпителиальные и мезангиальные иммунные комплексы (ИК), деструкция подоцита (Пд). БМ — базальная мембрана, МезК—мезангиальная клетка. × 10 000.
Почки часто поражаются при СКВ. Возникает два варианта гломерулонефрита: один с характерными морфологическими признаками — волчаночный нефрит, другой — без этих признаков, имеющий обычную картину гломеруло-нефрита (см. Болезни почек). При волчаночном нефрите почки увеличены, пестрые, с участками кровоизлияний (рис. 179). При микроскопическом исследовании волчаночный нефрит характеризуется наличием патологических изменений в ядрах (гематоксилиновые тельца), утолщением капиллярных мембран клубочков, принимающих вид проволочных петель, появлением гиалиновых тромбов и очагов фибриноидного некроза с фиксацией в них иммунных комплексов (рис. 180). В исходе волчаночного нефрита может возникать сморщивание почек с последующим развитием уремии.
Суставы вовлекаются в патологический процесс, однако изменения не достигают большой степени и редко сопровождаются деформацией (в таких случаях заболевание приобретает большое сходство с ревматоидным артритом). Гистологически в синовиальной оболочке выявляется клеточный инфильтрат, состоящий из макрофагов и плазматических клеток, встречаются склерозированные ворсинки, развиваются васкулиты. В околосуставной ткани наблюдаются участки мукоидного и фибриноидного набухания и поля склероза.
На коже боковых поверхностей лица симметрично появляются красные, слегка шелушащиеся участки, соединенные узкой красного цвета полосой на переносице (фигура бабочки). При обострении и прогрессировании болезни появляются высыпания и на других участках тела. С течением времени пятна приобретают коричневатый оттенок. При гистологическом исследовании в собственно коже в острых случаях видны отек и капилляриты, в артериолах наблюдаются фибриноидные изменения, вплоть до некроза. При затихании процесса в стенке сосудов и вокруг них появляются лимфоциты и макрофаги. Развиваются склероз, гиперкератоз, атрофия потовых и сальных желёз, что ведёт к облысению.
Осложнения. Наиболее опасные для жизни осложнения связаны с поражением почек — развитием их недостаточности на почве волчаночного нефрита. Иногда в связи с интенсивным лечением гормональными препаратами развиваются гнойные и септические процессы, «стероидный» туберкулез, а также эндокринные расстройства.
Смерть больных наступает чаще всего от почечной недостаточности или инфекции (сепсис, туберкулез).
Системная склеродермия (системный прогрессирующий склероз) — хроническое заболевание с преимущественным поражением соединительной ткани кожи и висцеральными проявлениями.
Этиология и патогенез. Предполагают, что основное значение в развитии заболевания имеет нарушение синтеза коллагена (аномальный неофибриллогенез), что показано при культивировании кожи больных системной склеродермией. Продукция несовершенного коллагена вызывает усиленный его распад и развитие фиброза. Не исключается роль вирусной инфекции (РНК-содержащий вирус) и генетических факторов. В патогенезе определенную роль могут играть аутоиммунные нарушения.
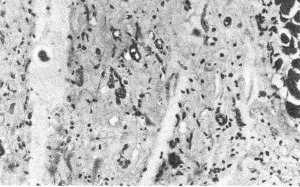
Рис. 181. Рубцовое поле в миокарде при системной склеродермии.
Патологическая анатомия. В коже и внутренних органах наблюдаются все виды дезорганизации соединительной ткани со слабовыраженной клеточной реакцией, заканчивающиеся грубым склерозом и гиалинозом. Кожа становится плотной и малоподвижной. В суставах отмечаются разной степени васкулиты, иногда с тромбами. Особенно опасно поражение сосудов почек в связи с возможностью развития некроза коркового слоя почек и острой их недостаточности — «истинная склеродермическая почка». Возможно преобладание крупноочагового кардиосклероза с сердечно-сосудистой недостаточностью — «склеродермическое сердце» (рис. 181) или фиброза базальных отделов лёгких и субплевральных областей — базальный пневмофиброз.
Осложнением у больных склеродермией наиболее часто является недостаточность тех органов и систем, в которых наиболее выражены склеротические изменения.
Узелковый периартериит см. Васкулиты.
Дерматомиозит — ревматическое заболевание, главным и ведущим клинико-морфологическим проявлением которого является системное поражение поперечнополосатой, в меньшей степени гладкой, мускулатуры и кожи. Наблюдаются случаи заболевания без поражения кожи, тогда его обозначают как полио-миозит. Дерматомиозит и полиомиозит встречаются в любом возрасте, преимущественно у женщин.
Этиология и патогенез. Предполагается вирусная природа заболевания. Косвенным подтверждением этого служит обнаружение у больных в цитоплазме эндотелио- и миоцитов тубулярных структур, сходных с парамиксовирусами. Показано значение генетической предрасположенности, описаны случаи семейного дерматомиозита. Развитие болезни связано, вероятнее всего, с нарушениями иммунологического гомеостаза и аутоиммунизацией. Пусковым механизмом является, видимо, вирусная инфекция. Очевидна связь дерматомиозита с опухолями, при этом опухолевые антигены могут быть перекрестно реагирующими с антигенами мышц, что усугубляет аутоиммунизацию. Отмечено улучшение состояния больных после удаления опухоли.
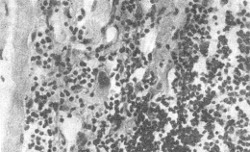
Рис. 182. Дерматомиозит. Дистрофические изменения мышечных волокон, клеточная инфильтрация межуточной ткани.
Патологическая анатомия. Наиболее часто изменения развиваются в скелетной мускулатуре, вмышцах глотки, гортани, диафрагмы, глазных мышцах. Мышцы становятся бледно-жёлтыми, отечными. В подкожной клетчатке, мышцах появляются очаги кальциноза. При микроскопическом исследовании постоянно обнаруживаются дистрофические изменения мышечных волокон, в них исчезает поперечная исчерченность, уменьшается содержание гликогена, резко понижается активность ряда ферментов. Многие мышечные волокна некротизированы, и в очагах некроза видно выпадение извести в виде мелких зерен. В соединительнотканной строме мышц, которая вовлекается в процесс вторично, развиваются отек и воспалительная реакция. В инфильтрате преобладают лимфоциты, макрофаги и плазматические клетки (рис. 182). Скопления лимфоцитов и макрофагов особенно выражены по ходу микрососудов, со стороны эндотелия капилляров отмечаются пролиферация и десквамация, вплоть до полного закрытия просвета.
Изменения внутренних органов при дерматомиозите постоянны. Они носят воспалительный, дистрофический или склеротический характер и наиболее часто наблюдаются в сердце, лёгких, желудочно-кишечном тракте.
Наиболее опасна пневмония, которая в детском возрасте часто является причиной гибели больного.
Лимфатические узлы и селезёнка обычно увеличены, с явлениями гиперплазии лимфоидной ткани и плазмоклеточной трансформацией.
Клинико-морфологические формы. Выделяют первичную (идиопатическую) и вторичную (опухолевую) формы дерматомиозита, морфологические проявления которых идентичны. Каждая из этих форм может иметь острое, подострое, непрерывно-рецидивирующее и хроническое течение.
Первичная форма обычно встречается у детей, вторичная — у взрослых.
Из опухолей, при которых развивается дерматомиозит, наиболее часто отмечается рак (яичников, молочной железы, лёгкого, желудка, кишечника). Нередко дерматомиозит служит первым проявлением опухоли.
138. Перечислите острые воспалительные заболевания легких: острый бронхит, острые пневмонии: этио-логия, классификация, патанатомия, особенности клинико-морфологических проявлений в зависимости от характера возбудителя.
Острые воспалительные заболевания легких имеют инфекционную природу, различные патогенез и клинико-морфоло-гические проявления, характеризуются развитием острого воспаления преимущественно в респираторных отделах легких. В отечественной литературе принято обозначать эти заболевания термином "острые пневмонии" [Абрикосов А.И., 1947; Струков А.И., Серов В.В., 1993; Замотаев И.П., 1989]. В зарубежных руководствах используются два термина "для обозначения острых воспалительных заболеваний легких — "пневмонии" и "пневмониты" [Creagh Т., Krauz Т., 1991]. К пневмониям относят только острые воспалительные заболевания легких бактериальной этиологии, основным морфологическим признаком которых является накопление экссудата в просветах альвеол. В тех случаях, когда острое воспаление преимущественно распространяется на альвеолярную стенку с вторич
ным накоплением экссудата в просветах альвеол, говорят об ин-терстициальном воспалении легких и обозначают его термином острый пневмонит. Таким образом, отечественные авторы придерживаются более широкого понимания термина "острые пневмонии", вбирающего в себя острое воспаление всего респираторного отдела легкого — паренхимы и интерстиция. При дальнейшем изложении материала будет использоваться данная концепция.
Эпидемиология . Острые пневмонии — одни из самых распространенных заболеваний с относительно высокой смертностью среди новорожденных и стариков. В России заболеваемость острыми пневмониями мало изменилась за последние 25 лет, несмотря на применение антибактериальной терапии, и составляет, по данным А.А.Роменского (1977), для крупозной пневмонии 0,9 %, для других пневмоний — 14,5 %. При пневмониях отмечается относительно высокая летальность, соответственно 1,2 и 0,7 %. В США острые пневмонии вместе с гриппом занимают третье место среди причин смерти и обнаруживаются у 3 % умерших.
Рис. 179. Почка при волчаночном нефрите.
Как уже говорилось, сосуды разного калибра подвергаются значительным изменениям, особенно сосуды микроциркуляторного русла — возникают артериолиты, капилляриты и венулиты. В стенке аорты в связи с поражением её микрососудов появляются вторичные изменения в виде эластолиза и мелких рубчиков в средней оболочке. В разных органах васкулиты вызывают вторичные изменения — дистрофию паренхиматозных элементов, некроз.
Рис. 180. Волчаночный нефрит. а — кариорексис, гиалиновые тромбы (гистологическая картина); б — субэпителиальные и мезангиальные иммунные комплексы (ИК), деструкция подоцита (Пд). БМ — базальная мембрана, МезК—мезангиальная клетка. × 10 000.
Почки часто поражаются при СКВ. Возникает два варианта гломерулонефрита: один с характерными морфологическими признаками — волчаночный нефрит, другой — без этих признаков, имеющий обычную картину гломеруло-нефрита (см. Болезни почек). При волчаночном нефрите почки увеличены, пестрые, с участками кровоизлияний (рис. 179). При микроскопическом исследовании волчаночный нефрит характеризуется наличием патологических изменений в ядрах (гематоксилиновые тельца), утолщением капиллярных мембран клубочков, принимающих вид проволочных петель, появлением гиалиновых тромбов и очагов фибриноидного некроза с фиксацией в них иммунных комплексов (рис. 180). В исходе волчаночного нефрита может возникать сморщивание почек с последующим развитием уремии.
Суставы вовлекаются в патологический процесс, однако изменения не достигают большой степени и редко сопровождаются деформацией (в таких случаях заболевание приобретает большое сходство с ревматоидным артритом). Гистологически в синовиальной оболочке выявляется клеточный инфильтрат, состоящий из макрофагов и плазматических клеток, встречаются склерозированные ворсинки, развиваются васкулиты. В околосуставной ткани наблюдаются участки мукоидного и фибриноидного набухания и поля склероза.
На коже боковых поверхностей лица симметрично появляются красные, слегка шелушащиеся участки, соединенные узкой красного цвета полосой на переносице (фигура бабочки). При обострении и прогрессировании болезни появляются высыпания и на других участках тела. С течением времени пятна приобретают коричневатый оттенок. При гистологическом исследовании в собственно коже в острых случаях видны отек и капилляриты, в артериолах наблюдаются фибриноидные изменения, вплоть до некроза. При затихании процесса в стенке сосудов и вокруг них появляются лимфоциты и макрофаги. Развиваются склероз, гиперкератоз, атрофия потовых и сальных желёз, что ведёт к облысению.
Осложнения. Наиболее опасные для жизни осложнения связаны с поражением почек — развитием их недостаточности на почве волчаночного нефрита. Иногда в связи с интенсивным лечением гормональными препаратами развиваются гнойные и септические процессы, «стероидный» туберкулез, а также эндокринные расстройства.
Смерть больных наступает чаще всего от почечной недостаточности или инфекции (сепсис, туберкулез).
СИСТЕМНАЯ СКЛЕРОДЕРМИЯ
Системная склеродермия (системный прогрессирующий склероз) — хроническое заболевание с преимущественным поражением соединительной ткани кожи и висцеральными проявлениями.
Этиология и патогенез. Предполагают, что основное значение в развитии заболевания имеет нарушение синтеза коллагена (аномальный неофибриллогенез), что показано при культивировании кожи больных системной склеродермией. Продукция несовершенного коллагена вызывает усиленный его распад и развитие фиброза. Не исключается роль вирусной инфекции (РНК-содержащий вирус) и генетических факторов. В патогенезе определенную роль могут играть аутоиммунные нарушения.
Рис. 181. Рубцовое поле в миокарде при системной склеродермии.
Патологическая анатомия. В коже и внутренних органах наблюдаются все виды дезорганизации соединительной ткани со слабовыраженной клеточной реакцией, заканчивающиеся грубым склерозом и гиалинозом. Кожа становится плотной и малоподвижной. В суставах отмечаются разной степени васкулиты, иногда с тромбами. Особенно опасно поражение сосудов почек в связи с возможностью развития некроза коркового слоя почек и острой их недостаточности — «истинная склеродермическая почка». Возможно преобладание крупноочагового кардиосклероза с сердечно-сосудистой недостаточностью — «склеродермическое сердце» (рис. 181) или фиброза базальных отделов лёгких и субплевральных областей — базальный пневмофиброз.
Осложнением у больных склеродермией наиболее часто является недостаточность тех органов и систем, в которых наиболее выражены склеротические изменения.
УЗЕЛКОВЫЙ ПЕРИАРТЕРИИТ
Узелковый периартериит см. Васкулиты.
ДЕРМАТОМИОЗИТ
Дерматомиозит — ревматическое заболевание, главным и ведущим клинико-морфологическим проявлением которого является системное поражение поперечнополосатой, в меньшей степени гладкой, мускулатуры и кожи. Наблюдаются случаи заболевания без поражения кожи, тогда его обозначают как полио-миозит. Дерматомиозит и полиомиозит встречаются в любом возрасте, преимущественно у женщин.
Этиология и патогенез. Предполагается вирусная природа заболевания. Косвенным подтверждением этого служит обнаружение у больных в цитоплазме эндотелио- и миоцитов тубулярных структур, сходных с парамиксовирусами. Показано значение генетической предрасположенности, описаны случаи семейного дерматомиозита. Развитие болезни связано, вероятнее всего, с нарушениями иммунологического гомеостаза и аутоиммунизацией. Пусковым механизмом является, видимо, вирусная инфекция. Очевидна связь дерматомиозита с опухолями, при этом опухолевые антигены могут быть перекрестно реагирующими с антигенами мышц, что усугубляет аутоиммунизацию. Отмечено улучшение состояния больных после удаления опухоли.
Рис. 182. Дерматомиозит. Дистрофические изменения мышечных волокон, клеточная инфильтрация межуточной ткани.
Патологическая анатомия. Наиболее часто изменения развиваются в скелетной мускулатуре, вмышцах глотки, гортани, диафрагмы, глазных мышцах. Мышцы становятся бледно-жёлтыми, отечными. В подкожной клетчатке, мышцах появляются очаги кальциноза. При микроскопическом исследовании постоянно обнаруживаются дистрофические изменения мышечных волокон, в них исчезает поперечная исчерченность, уменьшается содержание гликогена, резко понижается активность ряда ферментов. Многие мышечные волокна некротизированы, и в очагах некроза видно выпадение извести в виде мелких зерен. В соединительнотканной строме мышц, которая вовлекается в процесс вторично, развиваются отек и воспалительная реакция. В инфильтрате преобладают лимфоциты, макрофаги и плазматические клетки (рис. 182). Скопления лимфоцитов и макрофагов особенно выражены по ходу микрососудов, со стороны эндотелия капилляров отмечаются пролиферация и десквамация, вплоть до полного закрытия просвета.
Изменения внутренних органов при дерматомиозите постоянны. Они носят воспалительный, дистрофический или склеротический характер и наиболее часто наблюдаются в сердце, лёгких, желудочно-кишечном тракте.
Наиболее опасна пневмония, которая в детском возрасте часто является причиной гибели больного.
Лимфатические узлы и селезёнка обычно увеличены, с явлениями гиперплазии лимфоидной ткани и плазмоклеточной трансформацией.
Клинико-морфологические формы. Выделяют первичную (идиопатическую) и вторичную (опухолевую) формы дерматомиозита, морфологические проявления которых идентичны. Каждая из этих форм может иметь острое, подострое, непрерывно-рецидивирующее и хроническое течение.
Первичная форма обычно встречается у детей, вторичная — у взрослых.
Из опухолей, при которых развивается дерматомиозит, наиболее часто отмечается рак (яичников, молочной железы, лёгкого, желудка, кишечника). Нередко дерматомиозит служит первым проявлением опухоли.
138. Перечислите острые воспалительные заболевания легких: острый бронхит, острые пневмонии: этио-логия, классификация, патанатомия, особенности клинико-морфологических проявлений в зависимости от характера возбудителя.
Острые воспалительные заболевания легких имеют инфекционную природу, различные патогенез и клинико-морфоло-гические проявления, характеризуются развитием острого воспаления преимущественно в респираторных отделах легких. В отечественной литературе принято обозначать эти заболевания термином "острые пневмонии" [Абрикосов А.И., 1947; Струков А.И., Серов В.В., 1993; Замотаев И.П., 1989]. В зарубежных руководствах используются два термина "для обозначения острых воспалительных заболеваний легких — "пневмонии" и "пневмониты" [Creagh Т., Krauz Т., 1991]. К пневмониям относят только острые воспалительные заболевания легких бактериальной этиологии, основным морфологическим признаком которых является накопление экссудата в просветах альвеол. В тех случаях, когда острое воспаление преимущественно распространяется на альвеолярную стенку с вторич
ным накоплением экссудата в просветах альвеол, говорят об ин-терстициальном воспалении легких и обозначают его термином острый пневмонит. Таким образом, отечественные авторы придерживаются более широкого понимания термина "острые пневмонии", вбирающего в себя острое воспаление всего респираторного отдела легкого — паренхимы и интерстиция. При дальнейшем изложении материала будет использоваться данная концепция.
Эпидемиология . Острые пневмонии — одни из самых распространенных заболеваний с относительно высокой смертностью среди новорожденных и стариков. В России заболеваемость острыми пневмониями мало изменилась за последние 25 лет, несмотря на применение антибактериальной терапии, и составляет, по данным А.А.Роменского (1977), для крупозной пневмонии 0,9 %, для других пневмоний — 14,5 %. При пневмониях отмечается относительно высокая летальность, соответственно 1,2 и 0,7 %. В США острые пневмонии вместе с гриппом занимают третье место среди причин смерти и обнаруживаются у 3 % умерших.