Файл: Изучение молекулярных предикторов сосудистого риска опосредованного эндотелиальной дисфункцией у людей с сахарным диабетом 2 типа.docx
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 05.12.2023
Просмотров: 114
Скачиваний: 1
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
СОДЕРЖАНИЕ
КЛИНИЧЕСКАЯ ДИАГНОСТИКА ИНСУЛИНОВОЙ РЕЗИСТЕНТНОСТИ
МОЛЕКУЛЯРНЫЕ МАРКЕРЫ ИНСУЛИНОВОЙ РЕЗИСТЕНТНОСТИ
ШАПЕРОНЫ И МАЛЫЕ БЕЛКИ ТЕПЛОВОГО ШОКА
ИНСУЛИНОВАЯ РЕЗИСТЕНТНОСТЬ, КАРБОНИЛЬНЫЙ СТРЕСС И ДИСФУНКЦИЯ ЭНДОТЕЛИЯ
ИНСУЛИНОВАЯ РЕЗИСТЕНТНОСТЬ, АДИПОГЕННАЯ ДИФФЕРЕНЦИРОВКА И ТРАНСКРИПЦИОННЫЙ ФАКТОР PREP1
ИНСУЛИНОВАЯ РЕЗИСТЕНТНОСТЬ, ЛИПОДИСТРОФИЯ И МУТАЦИИ ГЕНОВ
ИНСУЛИНОВАЯ РЕЗИСТЕНТНОСТЬ И ГОРМОНЫ ИНКРЕТИНОВОГО РЯДА
Нарушение секреции инкретиновых гормонов и сниженный инкретиновый эффект являются причинами прогрессирования гипергликемии у пациентов с классическим СД2. Стимуляция выработки гормонов инкретинового ряда, в частности глюкагоноподобного пептида-1 (ГПП-1), ведет к компенсации углеводного обмена [54, 55]. Воздействие на этот механизм реализуется при назначении инкретин-направленной терапии [56, 57].
Для того чтобы проверить гипотезу о возможной роли гормонов инкретинового ряда в ремиссии СД2, было проведено клиническое исследование, в которое были включены пациенты с длительным анамнезом СД2 и ожирения (более 10–15 лет), получавшие терапию агонистом рецептора ГПП-1 лираглутидом в дозе, имитирующей максимальный инкретиновый эффект. Через 3–4 мес после получения препарата у пациентов было отмечено среднее снижение массы тела на 6,2 кг, гликированного гемоглобина крови (HbA1c) на 1,1% и повышение М-индекса с 1,74 мг/кг/мин до 2,52 мг/кг/мин, что говорит о снижении выраженности ИР и перспективах использования инкретин-направленной терапии для коррекции ИР у больных с СД2.
ЗАКЛЮЧЕНИЕ
Общая последовательность развития событий в патогенезе СД2 довольно понятна (рис. 2). Первичные факторы риска, включающие ожирение, воспаление и стресс различной природы, ведут к развитию ИР в клетках-мишенях инсулина. В классическом варианте в адипоцитах жировой ткани все факторы риска ИР действуют через единый механизм, связанный с латентным воспалением и хронической активацией стресс-зависимых киназ, таких как JNK и IKK. Они фосфорилируют субстрат инсулинового рецептора IRS, нарушая активацию инсулинового каскада и выход глюкозного транспортера Glut4 на поверхность клеток (см. рис. 1В). На молекулярном уровне ИР проявляется в снижении инсулинзависимого фосфорилирования компонентов инсулинового каскада, киназы Akt и белка AS160 (см. рис. 1Г). Наши данные подтверждают, что сайт-специфичное фосфорилирование Akt, AS160 и JNK может служить молекулярным маркером ИР в адипоцитах.
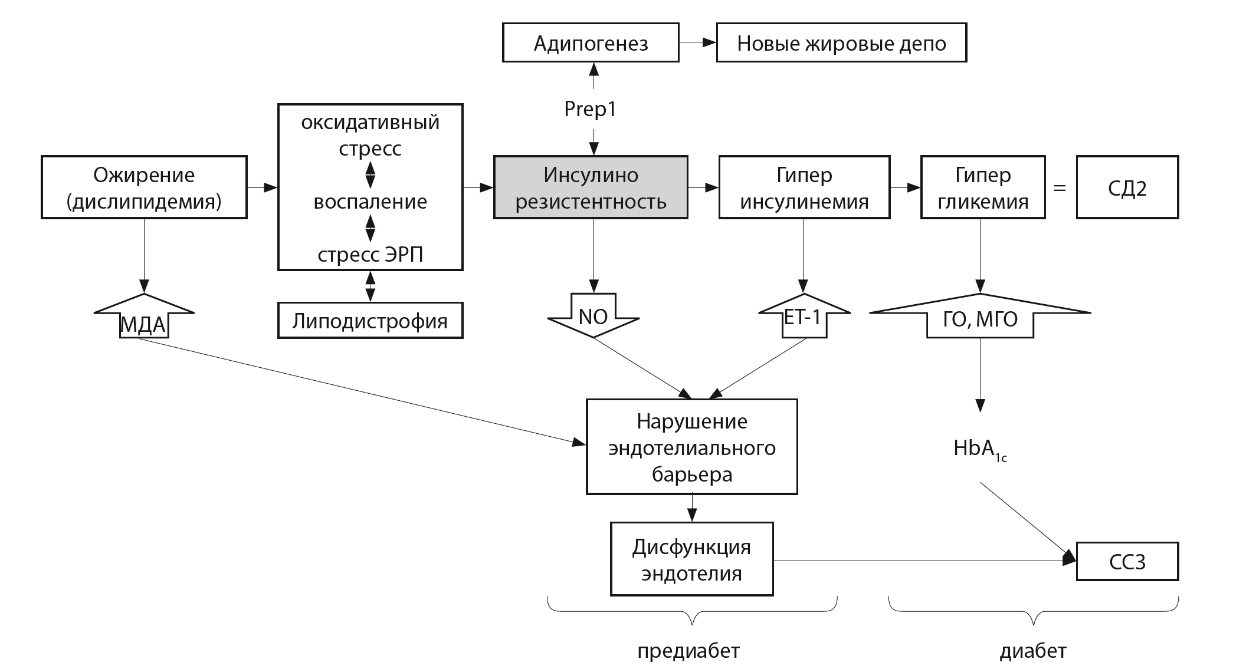
Рис. 2. Общая схема патогенеза сахарного диабета 2 типа и ассоциированных сердечно-сосудистых осложнений.
Липодистрофия представляет неклассический вариант патогенеза СД2, по крайней мере, в отношении жировой ткани. Наши и другие исследования определяют четкую связь между липодистрофией и ИР, но причинно-следственные взаимоотношения и патогенез СД2 при липодистрофии остаются малопонятными и требуют отдельных исследований.
Избыток свободных жирных кислот вследствие ожирения является важной причиной ИР и карбонильного стресса, повышая риск дисфункции сосудистого эндотелия и сердечно-сосудистых осложнений уже на первых стадиях патогенеза СД2. Активируя толл-подобные рецепторы TLR4, СЖК запускают воспалительные каскады и развитие ИР в адипоцитах. Кроме того, в клетках СЖК подвергаются перекисному окислению с образованием МДА как одного из продуктов. Физически воздействуя на эндотелий и модифицируя ряд белков, МДА изменяет структуру цитоскелета и межклеточных контактов, нарушая барьерные свойства эндотелия и повышая риск развития отеков, гипертонических осложнений и ангиопатий. Прогрессирование патологического процесса ведет к устойчивой гипергликемии и карбонильному стрессу за счет активных продуктов распада глюкозы. ГО и МГО химически модифицируют другие белки плазмы и клеток крови, что согласуется с повышением HbA1c.
sHSP защищают клетки от окислительного стресса и апоптоза на первых этапах патогенеза СД2, препятствуя агрегации денатурированных белков и развитию стресса ЭПР. Вместе с тем sHSP являются мишенью карбонильного стресса при гипергликемии, что ведет к изменению их структуры и функциональных свойств, уменьшению шапероноподобной активности и защитного действия. Тем самым sHSP могут опосредовать обратную связь от конечных к начальным этапам патогенеза СД2, усиливая эффекты оксидативного стресса и стресса ЭПР.
Персистирующая ИР запускает отложенные, длительные адаптационные процессы в организме; они ведут к перестройке всего метаболизма в процессе развития СД2. Такие длительные изменения закрепляются на транскрипционном уровне, где ключевую роль играют различные транскрипционные факторы. При этом направление перестроек часто определяется совокупным воздействием регуляторов транскрипции и внешних факторов. Например, в условиях гипергликемии может усиливаться экспрессия Prep1 в миоцитах, что снижает чувствительность мышц к инсулину и утилизацию ими глюкозы, но выполняет защитную функцию. Аналогичную роль может играть Prep1 в адипоцитах, где он также ослабляет
чувствительность к инсулину и захват глюкозы, предотвращая жировую перегрузку адипоцитов. В этой связи важно, что Prep1 участвует в регуляции адипогенеза, то есть он также подавляет процесс формирования новых жировых депо. В совокупности системный эффект Prep1 можно расценивать как компенсаторное отключение инсулиновой зависимости клеток в условиях гиперинсулинемии и пищевой перегрузки. Дальнейшие исследования должны быть направлены на валидацию Prep1 как терапевтической биомишени в животных моделях и поиск способов направленного воздействия на его экспрессию в инсулинчувствительных тканях.
Очевидно, что патогенез СД2 гораздо более сложен, чем схематично показанный на рис. 2. Определение других его участников и механизмов, безусловно, требует продолжения исследований.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Финансирование исследования. Обзорная работа подготовлена в рамках реализации научной программы, поддержанной грантом Российского научного фонда (проект №14-35-00026).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Дедов И.И. – концепция исследования, редактирование и финальное утверждение рукописи; Ткачук В.А. – дизайн экспериментальной части исследования, анализ результатов, написание текста; Гусев Н.Б. – дизайн экспериментальной части исследования, анализ результатов, написание текста; Ширинский В.П. – дизайн экспериментальной части исследования, анализ результатов, написание текста; Воротников А.В. – дизайн экспериментальной части исследования, анализ результатов, написание текста; Кочегура Т.Н. – анализ клинико-экспериментальных результатов исследования, написание текста, редактирование рукописи; Майоров А.Ю. – разработка дизайна проекта, формирование групп пациентов, набор клинического материала, анализ и интерпретация результатов, написание текста; Шестакова М.В. – разработка дизайна клинической части исследования, анализ и интерпретация результатов, написание и редактирование текста.
Благодарности. Авторы приносят благодарность за участие в исследовании:
-
сотрудникам ФГБУ «Национальный медицинский исследовательский центр эндокринологии» Минздрава России – д.м.н., профессору Тюльпакову А.Н., д.м.н., профессору Кураевой Т.Л., к.м.н. Соркиной Е.Л., к.м.н. Шестаковой Е.А., Филиппову Ю.И., аспиранту Кокшаровой Е.О., аспиранту Склянику И.А., аспиранту Мишиной Е.Е., Горелышеву А.С.; -
сотрудникам Московского государственного университета имени М.В. Ломоносова – к.м.н. Акопян Ж.А., к.б.н. Егорову А.Д., к.б.н. Кулебякину К.Ю., Степановой А.В., к.б.н. Судницыной М.В., к.б.н. Дацкевич П.Н., к.б.н. Нефедовой В.В., Рыжавской А.С., аспиранту Мурановой Л.К., Герасимович Е.С.; -
сотрудникам ФГБУ «Национальный медицинский исследовательский центр кардиологии» Министерства здравоохранения Российской Федерации – к.ф-м.н. Пенькову Д.Н., к.б.н. Хапчаеву А.Ю.
Авторы выражают искреннюю благодарность д.м.н. Плехановой О.С. за помощь в подготовке рукописи.
Все авторы внесли существенный вклад в проведение исследования и подготовку статьи, прочли и одобрили финальную версию перед публикацией.
СПИСОК ЛИТЕРАТУРЫ
↑1. Ткачук В.А., Воротников А.В. Молекулярные механизмы развития резистентности к инсулину // Сахарный диабет. -2014. -Т. 17. - №2. -С. 29-40. [Tkachuk VA, Vorotnikov AV. Molecular Mechanisms of Insulin Resistance Development. Diabetes mellitus. 2014;17(2):29-40. (In Russ.)] https://doi.org/10.14341/DM2014229-40
↑2. Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12(1):15-28. https://doi.org/10.1038/nrendo.2015.189
↑3. Stafeev IS, Menshikov MY, Tsokolaeva ZI, et al. Molecular Mechanisms of Latent Inflammation in Metabolic Syndrome. Possible Role of Sirtuins and Peroxisome Proliferator-Activated Receptor Type gamma. Biochemistry (Mosc). 2015;80(10):1217-1226. https://doi.org/10.1134/S0006297915100028
↑4. Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296(4):E581-591. https://doi.org/10.1152/ajpendo.90437.2008
↑5. Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55 Suppl 2:S9-S15. https://doi.org/10.2337/db06-S002
↑6. Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375(9733):2267-2277. https://doi.org/10.1016/s0140-6736(10)60408-4
↑7. Liu YF, Herschkovitz A, Boura-Halfon S, et al. Serine phosphorylation proximal to its phosphotyrosine binding domain inhibits insulin receptor substrate 1 function and promotes insulin resistance. Mol Cell Biol. 2004;24(21):9668-9681. https://doi.org/10.1128/MCB.24.21.9668-9681.2004
↑8. Zick Y. Uncoupling insulin signalling by serine/threonine phosphorylation: a molecular basis for insulin resistance. Biochem Soc Trans. 2004;32(Pt 5):812-816. https://doi.org/10.1042/BST0320812
↑9. Hojlund K. Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance. Dan Med J. 2014;61(7):B4890.
↑10. Stafeev IS, Vorotnikov AV, Ratner EI, et al. Latent Inflammation and Insulin Resistance in Adipose Tissue. Int J Endocrinol. 2017;2017:5076732. https://doi.org/10.1155/2017/5076732
↑11. Toth AE, Toth A, Walter FR, et al. Compounds blocking methylglyoxal-induced protein modification and brain endothelial injury. Arch Med Res. 2014;45(8):753-764. https://doi.org/10.1016/j.arcmed.2014.10.009
↑12. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42-61. https://doi.org/10.1210/er.2007-0015
↑13. Mitra A, Basak T, Datta K, et al. Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013;4:e582. https://doi.org/10.1038/cddis.2013.114
↑14. Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91(4):1123-1159. https://doi.org/10.1152/physrev.00023.2010
↑15. Kumano M, Furukawa J, Shiota M, et al. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol Cancer Ther. 2012;11(8):1661-1671. https://doi.org/10.1158/1535-7163.MCT-12-0072
↑16. Simar D, Jacques A, Caillaud C. Heat shock proteins induction reduces stress kinases activation, potentially improving insulin signalling in monocytes from obese subjects. Cell Stress Chaperones. 2012;17(5):615-621. https://doi.org/10.1007/s12192-012-0336-4
↑17. van Heijst JW, Niessen HW, Musters RJ, et al. Argpyrimidine-modified Heat shock protein 27 in human non-small cell lung cancer: a possible mechanism for evasion of apoptosis. Cancer Lett. 2006;241(2):309-319. https://doi.org/10.1016/j.canlet.2005.10.042