ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 19.11.2021
Просмотров: 3123
Скачиваний: 169
Иммуносупрессивная
терапии
показана
при
быстропрогрессирующих
вариантах
ГН
и
является
ургентной
терапией
.
Гистологическая
структура
ГН
широко
варьирует
у
детей
в
различных
странах
.
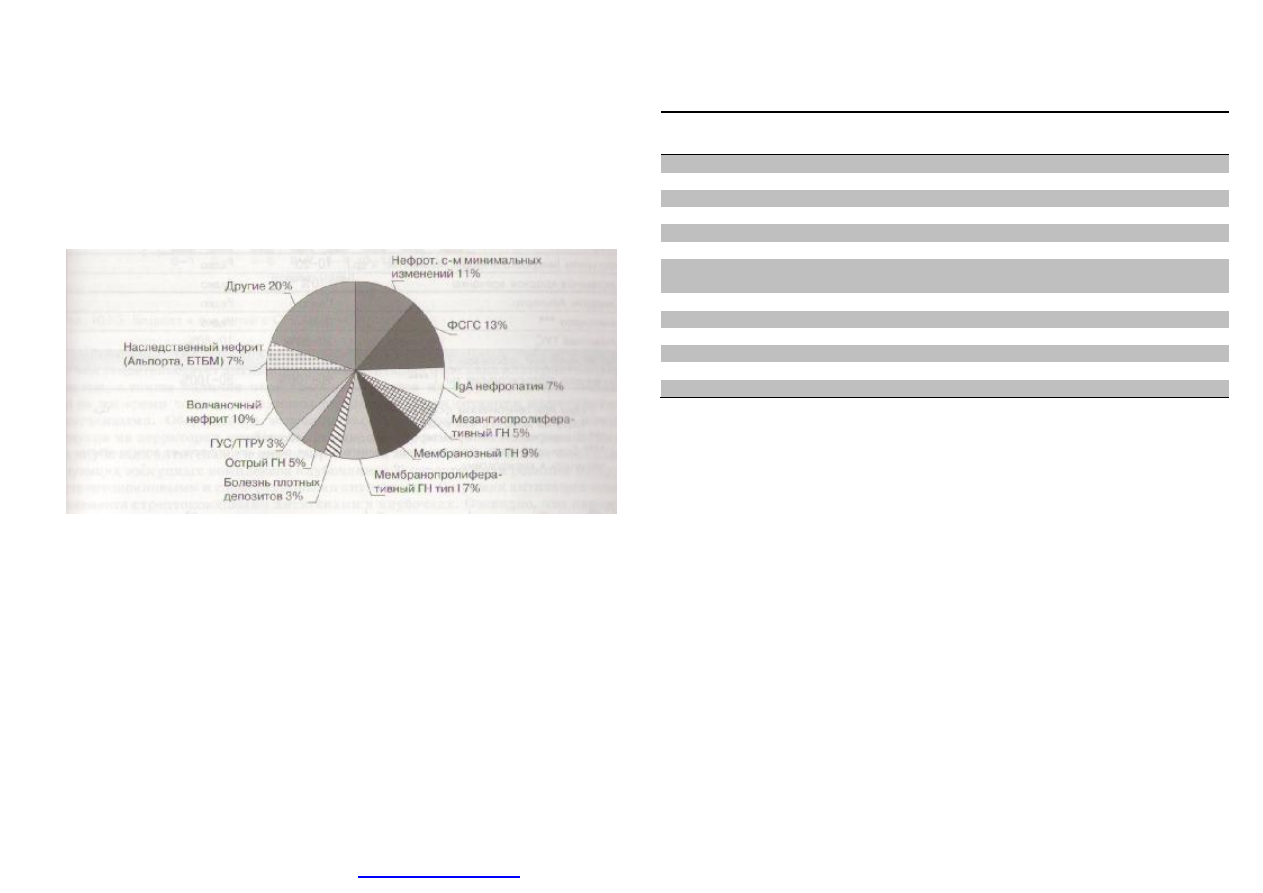
На
рис
. 10.1.2
представлена
частота
обнаружения
различных
форм
ГН
у
107
детей
,
обследованных
в
центре
детской
нефрологии
Армении
в
1992 -2008
гг
.
В
серию
не
вошел
51 (23%)
пациент
с
амилоидозом
почек
в
результате
периодической
болезни
—
эндемического
заболевания
в
Армении
.
Следует
учесть
,
что
малое
число
больных
острым
ГН
и
нефротическим
синдромом
минимальных
изменений
не
отражает
истинной
картины
распространенности
,
поскольку
менее
чем
5%
этих
пациентов
нуждались
в
проведении
нефробиопсии
.
Диагноз
у
остальных
был
основан
на
клинических
данных
.
Рис
. 10.1.2.
Частота
обнаружения
различных
форм
гломерулярных
болезней
у
167
детей
,
обследованных
в
центре
детской
нефрологии
Армении
в
1992-2008
гг
.
Возвратное
поражения
почек
после
трансплантации
В
табл
. 10.1.3
представлены
основные
заболевания
,
которые
имеют
тенденцию
к
рецидивированию
в
почечном
трансплантате
.
В
основном
это
гломерулярные
заболевания
.
Даже
тот
клиницист
,
который
непосредственно
не
занимается
почечной
трансплантацией
,
должен
знать
это
по
двум
причинам
:
1)
из
таблицы
видно
,
что
риск
развития
возврата
заболевания
существенно
зависит
от
характера
первичного
заболевания
,
поэтому
очень
важно
иметь
точный
диагноз
—
хронический
ГН
не
является
диагнозом
;
2)
большинство
заболеваний
возвращаются
несмотря
на
мощную
и
длительную
иммуносупрессию
,
предназначенную
для
профилактики
отторжения
.
Этот
факт
четко
демонстрирует
ограниченную
эффективность
иммупосупрессивных
препаратов
при
многих
гломерулярных
заболеваниях
(
в
том
числе
и
в
нативной
почке
еще
до
трансплантации
).
147
Таблица
10.1.3.
Частота
возврата
заболеваний
и
их
влияние
на
выживание
почечного
трансплантата
Заболевание
Вероятность
возврата
Вероятность
потери
трансплантата
при
возврате
Первичный
ФСГС
*
20-50%
40-60%
Мембранопролиферативный
ГН
(
тип
1)
30-70%
20-40%
Болезнь
плотных
депозитов
80-100
25-60%
Мембранозная
нефропатия
**
<10%
До
50%
IgA-
нефропатия
30-40%
10%
Нефрит
Шенлейна
—
Геноха
30-100%
10-20%
Васкулиты
(
микроскопический
полиангиит
и
др
.)
10-20
Редко
Системная
красная
волчанка
<10%
Редко
Синдром
Альпорта
редко
Редко
Амилоидоз
***
25%
Редко
Атипичный
ГУС
20-80%
10-80%
Метаболические
заболевания
Первичная
гипероксалурия
тип
1 ****
90-100%
80-100%
*
Редко
при
генетических
формах
.
**
Чаще
de novo
ГН
,
чем
возвратный
.
***
В
зависимости
от
персистирования
основного
заболевания
.
****
Если
проведена
изолированная
трансплантация
почки
или
отсутствует
полная
чувствительность
к
пиридоксину
.
2.
Острый
постинфекционный
(
постстрептококковый
)
гломерулонефрит
Острый
гломерулонефрит
характеризуется
внезапным
началом
и
типичными
проявлениями
в
виде
мочевого
синдрома
(
гематурия
,
протеинурия
)
и
,
нередко
,
отеков
,
гипертензии
и
нарушения
функции
почек
.
Хотя
поражение
почек
может
ассоциироваться
с
различными
инфекциями
(
бактериальный
эндокардит
,
стафилококк
,
вирусы
и
др
.),
прототипом
заболевания
является
острый
постстрептококковый
ГН
(
ОПСГН
).
Связь
между
стрептококковой
инфекцией
(
скарлатина
)
и
поражением
почек
была
распознана
еще
в
XVIII
столетии
.
Этиология
и
эпидемиология
.
Имеется
отчетливая
связь
распространенности
ОПСГН
с
социально
-
экономическим
состоянием
общества
.
Заболевание
обычно
носит
спорадический
характер
,
однако
нередки
вспышки
,
обусловленные
резким
снижением
уровня
жизни
,
например
в
некоторых
странах
бывшего
СССР
в
начале
1990-
х
гг
.
ОПСГН
обычно
начинается
через
1-3
недели
после
респираторной
либо
кожной
инфекции
,
вызванной
«
нефритогенными
»
штаммами
в
-
гемолитического
стрептококка
группы
А
.
Фактор
(
ы
),
обусловливающие
нефритогенность
,
остаются
недостаточно
изученными
.
148
PDF created with pdfFactory trial version

ОПСГН
обычно
поражает
детей
школьного
возраста
;
мальчики
болеют
вдвое
чаще
,
чем
девочки
(
рис
. 10.1.3).
Рис
. 10.1.3.
Возраст
и
пол
детей
с
ОПСГН
(
п
=474)
Патогенез
.
ОПСГН
является
иммунно
-
опосредованным
заболеванием
,
о
чем
свидетельствует
активация
комплементарного
каскада
альтернативным
путем
,
а
также
наличие
иммунных
комплексов
на
территории
клубочков
.
В
то
же
время
точные
механизмы
поражения
почек
остаются
недостаточно
изученными
.
Обсуждается
возможность
: 1)
образования
иммунных
комплексов
на
территории
клубочков
(in situ)
между
ранее
имплантированными
в
клубочках
антигенами
и
циркулирующими
антителами
; 2)
захват
циркулирующих
иммунных
комплексов
клубочками
; 3)
перекрестная
реакция
между
стрептококковыми
и
гломерулярными
антигенами
; 4)
прямая
активации
комплемента
стрептококковыми
антигенами
в
клубочках
.
Очевидно
,
что
наряду
с
инфекцией
в
развитии
заболевания
существенную
роль
играют
особенности
иммунного
ответа
.
Клиническая
картина
.
ОПСГН
характеризуется
внезапным
началом
гематурии
,
протеинурии
,
нередко
в
сочетании
с
отеками
и
гипертензией
через
1—3
недели
после
стрептококковой
инфекции
.
Хотя
клиническим
эквивалентом
заболевания
считается
острый
нефритический
синдром
,
у
большинства
пациентов
заболевание
ограничивается
мочевым
синдромом
.
Макрогематурия
отмечается
-40%
диагностированных
случаев
и
продолжается
до
2-
х
недель
.
Гематурия
сопровождается
умеренной
протеинурией
.
Нефротический
синдром
отмечается
крайне
редко
и
требует
дополнительного
гистологического
подтверждения
диагноза
ОПСГН
.
У
многих
пациентов
имеется
транзиторное
снижение
почечной
функции
в
той
или
иной
степени
с
развитием
олигурии
.
Анурия
и
почечная
недостаточность
,
требующая
замещения
функции
,
отмечаются
редко
.
Отеки
,
обычно
умеренные
,
и
гипертензия
являются
результатом
задержки
жидкости
и
соли
.
В
отдельных
случаях
развивается
застойная
сердечная
недостаточность
со
значительной
перегрузкой
легочной
циркуляции
,
проявляющейся
кашлем
,
респираторным
дистресс
-
синдромом
,
крепитацией
в
легких
,
сердечным
ритмом
галопа
.
Гипертензия
отмечается
у
большинства
пациентов
,
является
объемза
-
149
висимой
,
может
сопровождаться
головными
болями
,
тошнотой
,
рвотой
,
судорогами
(
гипертензионная
энцефалопатия
).
Указанные
осложнения
могут
представлять
опасность
для
жизни
.
Диагностика
заболевания
основана
на
анамнестических
,
бактериологических
и
серологических
данных
в
пользу
перенесенной
стрептококковой
инфекции
(
повышение
уровня
антител
к
стрептококковым
антигенам
:
аитистрептолизину
-0,
антигиалуронидазе
и
др
.),
результатах
физикального
обследования
(
отеки
,
гипертензии
),
лабораторных
показателях
(
умерен
ная
протеинурия
,
макро
-
и
микрогематурия
,
нарушение
функции
почек
,
активация
комплементарного
каскада
по
альтернативному
пути
),
характере
течения
заболевания
(
циклическое
).
Учитывая
цикличность
течения
ОПСГН
,
у
преобладающего
большинства
детей
необходимость
в
гистологическом
подтверждении
диагноза
возникает
очень
редко
,
в
основном
при
наличии
нехарактерных
симптомов
:
анурии
,
нефротическом
синдроме
,
длительной
(
более
6-8
мес
)
протеинурии
и
др
.
Гистологическая
картина
характеризуется
эндокапиллярным
пролиферативным
ГН
с
отложением
иммуноглобулинов
и
комплемента
на
территории
клубочков
.
На
электронной
микроскопии
определяются
субэпителиальные
депозиты
в
виде
горбиков
(humps).
Детальное
описание
приведено
в
главе
7.
Терапия
.
Постельный
режим
назначается
исключительно
в
случае
тяжелых
клинических
появлений
(
отек
легкого
и
др
.).
Терапия
острого
нефритического
синдрома
включает
в
себя
ограничение
жидкости
и
соли
,
назначение
петлевых
диуретиков
и
гипотензивных
средств
(
блокаторы
кальциевых
каналов
и
др
.).
Пациенты
с
отеком
легкого
и
гипертензионной
энцефалопатией
нуждаются
в
агрессивной
диуретической
и
антигипертензивной
терапии
.
Необходимость
в
ЗПТ
возникает
редко
(4
из
500
пациентов
).
Безотносительно
к
присутствию
стрептококковой
инфекции
в
момент
диагностики
ГН
,
во
многих
центрах
рутинно
назначается
антибиотик
пенициллинового
ряда
.
Профилактическую
антибактериальную
терапию
рекомендуется
проводить
членам
семьи
,
которые
находились
в
непосредственном
контакте
с
пациентом
,
особенно
в
условиях
вспышки
заболевания
.
В
случае
быстропрогрессирующего
течения
ОПСГН
и
наличии
многочисленных
полулуний
в
биоптате
,
необходимо
обсудить
возможность
назначения
пульс
-
терапии
метилпреднизолоном
и
цитотоксических
препаратов
,
эффективность
которых
остается
недоказанной
.
По
мере
ликвидации
отеков
гипертензии
и
др
.
ребенок
может
посещать
школу
.
Остаточная
микрогематурия
не
является
противопоказанием
для
спортивной
активности
.
Течение
и
прогноз
.
В
дебюте
ОПСГН
смертность
в
результате
отека
легкого
и
мозга
отмечается
у
менее
чем
1%
больных
.
У
преобладающего
большинства
пациентов
развивается
полная
ремиссия
.
Нефритический
синдром
продолжается
1-2
недели
,
тогда
же
исчезает
макрогематурия
.
Протеинурия
может
продолжаться
несколько
месяцев
,
а
микрогематурия
—
несколько
лет
.
После
нормализации
анализов
пациент
в
дальнейшем
наблюдении
не
нуждается
.
Весьма
редко
заболевание
принимает
прогрессирующий
характер
.
Неблагоприятными
факторами
в
дебюте
являются
нефротический
синдром
,
длительное
нарушение
функции
почек
,
PDF created with pdfFactory trial version

отдельные
гистологические
изменения
,
такие
как
многочисленные
полулуния
,
адгезии
,
склероз
.
Нередко
ОПСГН
клинически
невозможно
отличить
от
дебюта
МПГН
.
Нормализация
С
3
компонента
комплемента
через
3—6
месяцев
от
начала
заболевания
является
веским
аргументом
в
пользу
ОПСГН
.
Имеются
единичные
описания
повторных
эпизодов
заболевания
.
3.
Мембранопролиферативный
(
мезангиокапиллярный
)
гломерулонефрит
Мембранопролиферативный
ГН
(
МПГН
)
включает
в
себя
ряд
неоднородных
патологических
состояний
,
имеющих
морфологическое
сходство
,
но
различные
патогенетические
механизмы
развития
.
Гистологическая
картина
характеризуется
гломерулярной
гиперцеллюлярностью
,
расширением
мезангиального
матрикса
,
утолщением
капиллярной
стенки
.
Патогенетически
МПГН
изначально
отличается
от
других
вариантов
хронического
гломерулонефрита
наличием
у
большинства
пациентов
гипокомплементемии
.
Выделяют
первичный
(
идиопатический
)
и
вторичный
варианты
МПГН
.
Традиционно
первичпмй
МПГН
подразделяли
на
3
типа
,
в
первую
очередь
,
на
основании
ультраструктурного
исследования
почечной
ткани
.
В
настоящее
время
выделяют
МПГН
тип
I (
в
прошлом
тип
I +
тип
III)
и
болезнь
плотных
депозитов
(
БПД
в
прошлом
МПГН
тип
II) (
см
.
табл
. 10.1.4).
Более
того
,
при
исследовании
особенностей
патогенеза
,
характера
гистологических
изменений
,
течения
и
прогноза
(
в
том
числе
и
после
трансплантации
почки
)
накапливается
все
больше
свидетельств
того
,
что
МПГН
тип
I
и
БПД
следует
рассматривать
как
раздельные
нозологические
единицы
.
Гистологические
изменения
,
характерные
для
этих
заболеваний
,
подробно
изложены
в
главе
7.3.
Эпидемиология
.
В
литературе
имеются
весьма
скудные
данные
о
распространенности
МПГН
у
детей
,
которые
зависят
от
возраста
и
региона
.
Так
,
среди
детей
с
нефротическим
синдромом
Европы
и
Северной
Америки
удельный
вес
МПГН
составляет
6,2%,
в
то
время
как
эта
цифра
в
Азии
,
Африке
и
Южной
Америке
достигает
40%.
В
целом
,
заболеваемость
МПГН
в
экономически
развитых
странах
очень
низкая
,
в
то
время
как
она
остается
высокой
в
развивающихся
странах
.
Болеют
обычно
дети
старше
10
лет
и
подростки
.
Чаще
встречается
МПГН
тип
I.
БПД
является
редким
заболеванием
,
составляя
лишь
5%
всех
первичных
случаев
МПГН
.
Таблица
10.1.4.
Дифференциальная
диагностика
МПГН
тип
I,
БПД
и
МН
*
МПГН
1
БПД
МН
Комплемент
С
3
Часто
снижен
Всегда
снижен
В
норме
Этиология
Первичный
/
вторичны
й
Некоторые
варианты
генетические
Первичный
/
вторичны
й
Течение
Медленное
или
быстрое
прогрессирование
Часто
быстропрогрессирующи
й
Нередко
спонтанные
ремиссии
Возврат
после
трансплантаци
и
Часто
,
риск
потери
почки
Всегда
,
высокий
риск
потери
почки
Нечасто
,
но
может
развиься
de novo
МН
*
МН
–
мембранозная
нефропатия
151
МПГН
тип
I
Патогенез
.
МПГН
тип
I
развивается
в
результате
депозиции
циркулирующих
иммунных
комплексов
преимущественно
в
субэндотелиальном
пространстве
и
мезангии
.
Далее
происходит
активация
комплемента
по
классическому
пути
(
низкий
уровень
СЗ
и
С
4),
образование
мембраноатакующего
комплекса
(
МАК
) (
С
5-
Ь
9),
выброс
хемотактических
факторов
,
стимулирующих
накопление
лейкоцитов
и
тромбоцитов
в
клубочке
с
выбросом
цитокинов
и
факторов
роста
и
др
.,
приводящих
в
свою
очередь
к
пролиферации
мезангиальных
клеток
и
экспансии
мезангиального
матрикса
.
Клиническая
картина
.
Заболевание
проявляется
обычно
на
второй
декаде
жизни
.
У
50%
развивается
нефротический
синдром
,
у
20%
отмечается
макрогематурия
.
У
остальных
больных
ведущим
является
умеренная
протеинурия
.
Одна
треть
пациентов
в
дебюте
имеет
гипертензию
,
примерно
столько
же
-
почечную
недостаточность
.
Заболевание
является
прогрессирующим
.
Терминальная
почечная
недостаточность
может
развиться
уже
в
детском
возрасте
.
У
отдельных
пациентов
заболевание
протекает
по
типу
быстропрогрессирующего
ГН
.
Терапия
.
Длительное
(
более
1
года
)
лечение
преднизолоном
в
дозе
60
мг
/
м
2
альтернирующим
курсом
с
последующим
переходом
на
поддерживающие
дозы
,
может
быть
эффективным
в
отношении
снижения
уровня
протеинурии
и
почечной
выживаемости
.
Другими
препаратами
являются
мофетил
микфенолат
,
ритуксимаб
.
В
связи
с
небольшим
числом
наблюдений
каких
-
либо
выводов
в
отношении
их
эффективности
сделать
невозможно
.
Редким
пациентам
с
быстропрогрессирующим
курсом
заболевания
необходимо
назначение
пульс
-
терапии
метилпреднизолоном
и
цитотоксических
препаратов
.
Большинство
больных
н
|
уждается
лишь
в
поддерживающей
терапии
(
см
.
выше
).
Вторичный
МПГН
тип
1
МПГН
тип
1,
который
гистологически
не
отличается
от
первичных
форм
,
может
огмечаться
при
ряде
инфекций
(
гепатит
В
и
С
и
др
.),
системных
заболеваниях
(
криоглобулинемия
,
СКВ
и
др
.),
опухолях
и
др
.
Терапевтические
мероприятия
при
вторичном
МПГН
помимо
общих
мероприятий
,
направленных
на
снижение
уровня
прогрессирования
заболевания
,
включают
также
терапию
первичной
причины
(
инфекция
и
др
.)
Болезнь
плотных
депозитов
(
мембранопролиферативный
гломерулонефрит
,
тип
II, Dense Deposit Disease — DDD)
Болезнь
плотных
депозитов
(
БПД
)
является
редкой
формой
ГН
,
преимущественно
отмечается
в
детском
и
юношеском
возрастах
.
В
основе
названия
заболевания
лежит
характерная
электронно
-
микроскопическая
картина
:
наличие
электронно
-
плотного
материала
в
толще
гломерулярной
базальной
мембраны
(
ГБМ
).
Патогенез
.
БПД
является
комплемент
-
опосредованным
заболеванием
.
В
норме
альтернативный
путь
активируется
путем
самопроизвольного
расщепления
СЗ
-
фракции
.
В
результате
присоединения
к
СЗ
фактора
В
и
пропердина
образуется
СЗ
-
конвертаза
(
СЗЬВЬ
).
Этот
фермент
регулирует
весь
дальнейший
процесс
активации
до
образования
мембрано
-
атакующего
комплекса
. 152
PDF created with pdfFactory trial version

Активность
С
3
конвертазы
в
свою
очередь
регулируют
,
с
одной
стороны
,
стабилизирующие
аутоантитела
—
так
называемые
С
3-
нефритический
фактор
(
НФ
),
с
другой
—
фактор
Н
,
способствующий
разрушению
фермента
.
Присутствие
СЗНФ
и
/
или
дефицит
фактора
Н
(
мутация
гена
,
наличие
специфических
антител
и
др
.)
способствуют
бесконтрольной
активации
комплемента
по
альтернативному
пути
и
в
ряде
случаев
развитию
БПД
.
Большинство
пациентов
БПД
не
имеют
мутаций
гена
,
кодирующего
фактор
Н
,
однако
даже
небольшие
колебания
концентрации
этого
вещества
либо
дисфункция
являются
факторами
риска
.
Точные
механизмы
поражения
почек
,
в
частности
образования
характерных
плотных
депозитов
и
развития
клинической
симптоматики
,
остаются
неизученными
.
Клубочки
,
по
-
видимому
,
поражаются
в
результате
воздействия
хемотоксинов
лейкоцитов
и
МАК
.
Клиническая
картина
.
БПД
чаще
поражает
детей
и
молодых
лиц
обоих
полов
равномерно
.
Иногда
заболевание
диагностируется
у
взрослых
,
часть
из
которых
страдают
моноклональной
гаммопатией
.
Заболеванию
может
предшествовать
респираторная
инфекция
,
в
том
числе
стрептококковая
.
У
всех
пациентов
отмечаются
протеинурия
и
гематурия
(
макроскопическая
у
-23%).
По
данным
различных
авторов
,
нефритический
синдром
отмечается
у
16-38%,
нефротический
синдром
у
12-55%
пациентов
.
У
1/
з
детей
снижены
функции
почек
.
Заболевание
может
принять
быстропрогрессирующое
течение
.
К
экстраренальным
проявлениям
БПД
относятся
изменения
сетчатки
глаза
и
парциальная
липодистрофия
,
которые
также
являются
следствием
ннрушения
активации
комплемента
альтернативным
путем
.
Окулярные
изменения
характеризуются
появлением
характерных
бляшек
(drusen)
на
сетчатке
,
которые
могут
привести
к
потере
зрения
.
При
парциальной
липодистрофии
отмечается
исчезновение
подкожно
-
жировой
клетчатки
верхней
половины
тела
.
Диагноз
.
Предположить
заболевание
возможно
на
основании
клинических
признаков
ГН
(
мочевой
,
нефритический
,
нефротический
синдромы
,
снижение
функции
почек
),
постоянно
низком
уровне
СЗ
-
фракции
комплемента
,
обнаружении
типичных
экстраренальных
признаков
(
см
.
табл
. 10.1.4).
Однако
диагностика
БПД
основана
исключительно
на
данных
гистологического
исследования
почечной
ткани
(
в
особенности
—
электронной
микроскопии
).
При
этом
определяется
патогномоничный
признак
-—
отложение
электронно
-
плотного
материала
в
толще
ГБМ
,
которая
замещает
lamina densa (
см
.
главу
7).
Специальные
диагностические
тесты
у
пациента
и
у
членов
семьи
(
концентрации
и
активности
фактора
Н
и
др
.)
обычно
проводятся
с
научной
целью
лишь
после
гистологического
подтверждения
диагноза
.
Учитывая
разнообразие
клинических
появлений
,
дифференциальная
диагностика
до
проведения
нефробиопсии
включает
большинство
гломерулопатий
,
в
особенности
острый
постинфекционный
ГН
.
Лечение
.
Лечение
БПД
включает
в
себя
как
основные
мероприятия
по
сохранению
функции
почек
и
предотвращению
прогрессирования
заболевания
(
см
.
выше
),
так
и
специальные
методы
,
направленные
на
ликвидацию
нарушений
,
специфических
для
данного
заболевания
.
В
случае
дефицита
фактора
Н
эффективным
может
оказаться
его
возмещение
путем
вливания
свежезамороженной
плазмы
.
Наличие
СЗНФ
предполагает
проведение
плазмафереза
для
удаления
этих
антител
.
Иммуносупрессивная
терапия
обычно
неэффективна
,
за
исключением
пульс
терапии
метилпреднизолоном
при
наличии
полулунного
ГН
с
быстропрогрессиру
-
ющим
течением
.
В
литературе
имеются
единичные
сообщения
об
эффективности
биологических
препаратов
(
ритуксимаб
,
экулизумаб
и
др
.).
Следует
отметить
,
что
так
же
,
как
и
в
отношении
любого
редкого
заболевания
,
нет
работ
,
которые
могли
бы
в
достаточной
степени
оценить
эффективность
тех
или
иных
методов
лечения
.
Прогноз
.
В
целом
прогноз
БПД
неблагоприятный
.
Терминальная
почечная
недостаточность
развивается
у
70%
детей
в
среднем
в
течение
9
лет
.
Имеются
редкие
сообщения
о
развитии
полной
ремиссии
.
Трансплантация
(
табл
. 10.1.3
и
10.1.4).
Почти
во
всех
случаях
БПД
повторяется
в
трансплантированной
почке
,
чему
не
препятствует
стандартная
иммуносупрессия
против
отторжения
.
Учитывая
то
,
что
заболевание
при
этом
часто
бывает
менее
агрессивным
,
чем
в
дебюте
,
пациенты
с
БПД
должны
рассматриваться
как
кандидаты
для
проведения
трансплантации
почки
.
4.
Мембранозная
нефропатия
Мембранозная
нефропатия
(
МН
),
являясь
наиболее
частой
причиной
нефротического
синдрома
у
взрослых
,
у
детей
встречается
редко
и
составляет
1-2%
от
числа
пациентов
с
нефротическим
синдромом
.
МН
может
быть
первичной
(
идиопатической
)
либо
вторичной
по
отношению
к
аутоиммунным
(
СКВ
),
инфекционным
(
гепатиты
В
и
С
),
опухолевым
(
нейробластома
,
опухоль
Вильмса
)
заболеваниям
,
воздействию
отдельных
препаратов
.
Патогенез
.
Гистологические
изменения
при
МН
свидетельствуют
о
главенст
вующей
роли
субэпителиальной
(
эпимембранозной
)
депозиции
иммунных
комплексов
и
поражении
подоцитов
(
подробнее
в
главе
7).
Экспериментальная
модель
МН
у
крыс
(
Неу
mann nephritis)
с
активной
иммунизацией
компонентами
почечной
ткани
,
дала
возможность
достаточно
полно
изучить
патогенез
заболевания
.
Образовавшиеся
антитела
проникают
через
клубочковый
фильтр
,
связываются
с
антигеном
,
образуя
субэпителиальные
иммунные
комплексы
in situ.
Сам
антиген
у
человека
неизвестен
,
возможным
кандидатом
является
нейтральная
эндопептидаза
подоцитов
(NEP).
Недавно
были
идентифицированы
мутации
гена
,
кодирующего
эндопептидазу
(
ММЕ
).
Большинство
антигенов
при
вторичной
МН
остаются
неизвестными
.
Тем
не
менее
HBs
и
НВе
антигены
рвпатита
В
были
идентифицированы
в
иммунных
комплексах
.
Клиническая
картина
.
В
60—70%
случаях
в
дебюте
отмечается
нефротический
синдром
,
у
остальных
—
протеинурия
,
которая
в
большинстве
случаев
в
течение
1-2
лет
прогрессирует
до
развития
нефротического
синдрома
.
У
30-40%
больных
отмечается
также
микрогематурия
.
Макрогематурия
и
эритроцитарные
цилиндры
,
ОПН
встречаются
крайне
редко
и
требуют
проведения
дифференциальной
диагностики
с
другими
заболеваниями
.
К
началу
МН
преобладающее
большинство
пациентов
имеет
нормальное
АД
.
Гипертензия
по
ходу
заболевания
отмечается
лишь
в
10-20%
случаев
.
Стандартная
гормональная
терапия
обычно
неэффективна
.
Течение
.
МН
является
хроническим
заболеванием
,
для
которого
характерны
спонтанные
ремиссии
и
рецидивы
.
В
связи
с
редкостью
патологии
у
детей
болезнь
наиболее
полно
изучена
у
взрослых
.
По
данным
различных
авторов
,
спонтанная
PDF created with pdfFactory trial version
ремиссия
в
течение
5
лет
отмечена
у
20-40%
пациентов
.
Остальные
пациенты
(-
п
o
30%)
либо
уходят
в
почечную
недостаточность
,
либо
поддерживают
нормальную
функцию
почек
на
фоне
активного
заболевания
. 10-
летняя
почечная
выживаемость
составляет
70%.
Отмечено
,
что
у
детей
чаще
,
чем
у
взрослых
,
имеют
место
макрогематурия
и
рецидивы
;
реже
—
тромбоэмболические
осложнения
.
В
целом
,
прогноз
у
детей
более
благоприятный
.
Течение
вторичных
форм
МН
менее
изучено
.
У
30—60%
пациентов
с
HBV-
ассоциированной
МН
развивается
спонтанная
ремиссия
.
У
остальных
—
сохраняется
протеинурия
и
/
или
нефротический
синдром
.
Дифференциальная
диагностика
между
МПГН
тип
I,
БПД
и
МН
приведена
в
табл
. 10.1.4.
Терапия
.
В
связи
с
редкостью
заболевания
у
детей
данные
литературы
в
основном
касаются
взрослых
.
В
течение
6
месяцев
от
начала
заболевания
пациенты
должны
получать
исключительно
поддерживающую
терапию
,
поскольку
за
этот
период
у
значительного
числа
из
них
может
развиться
спонтанная
ремиссия
.
Анализ
существующих
исследований
показал
,
что
гормональная
монотерапия
не
увеличивает
возможности
ремиссий
и
не
повышает
показатель
5-
летней
почечной
выживаемости
,
по
сравнению
с
поддерживающей
терапией
.
Имеются
данные
об
эффективности
циклоспорина
.
Роль
иммуносупрессивной
терапии
у
детей
оценена
неоднозначно
и
основана
на
малочисленных
наблюдениях
.
Как
бы
то
ни
было
,
возможность
назначения
той
или
иной
терапии
должна
быть
рассмотрена
только
у
детей
с
персистирующим
нефротическим
синдромом
.
5.
Иммуноглобулин
А
-
нефропатия
и
нефрит
Шенлейна
—
Геноха
Иммуноглобулин
А
(IgA)-
нефропатия
и
нефрит
Шенлейна
—
Геноха
(
ШГ
)
имеют
весьма
схожую
картину
поражения
почек
с
преимущественно
мезангиальной
пролиферацией
и
наличием
иммунных
комплексов
,
содержащих
IgA.
Эти
изменения
могут
быть
в
составе
системного
васкулита
(
нефрит
ШГ
)
либо
ограниченны
почечным
поражением
(IgA-
нефропатия
) (
см
.
табл
. 10.1.5).
Иммуноглобулин
А
(IgA)
-
нефропатия
является
хроническим
заболеванием
.
Она
может
проявиться
в
любом
возрасте
,
однако
наиболее
часто
встречается
на
второй
и
третьей
декадах
жизни
,
вдвое
чаще
у
лиц
мужского
пола
.
Заболевание
может
быть
исключительно
диагностировано
на
основании
гистологического
исследования
почечной
ткани
с
выявлением
мезангиальной
депозиции
IgA,
которую
впервые
продемонстрировал
парижский
врач
Берже
в
1968
г
. (Berger's disease).
Поскольку
IgA-
нефропатия
во
многих
случаях
протекает
латентно
,
частота
ее
обнаружения
во
многом
зависит
от
таких
факторов
,
как
наличие
государственных
скрининговых
программ
исследования
мочи
,
доступность
и
показания
к
нефробиопсии
(
при
изолированной
микрогематурии
).
Очевидно
,
что
имеется
значительное
число
недиагностированных
случаев
.
Патогенез
.
Характерным
является
отложение
IgA,
часто
в
сочетании
с
IgG
и
СЗ
,
преимущественно
в
мезангии
и
,
в
меньшей
степени
,
по
ходу
ГБМ
.
Гистологическая
классификация
IgA-
нефропатии
приведена
в
главе
7
и
основана
на
наличии
или
степени
пролиферации
мезангиальных
клеток
.
Несмотря
на
усилия
многочисленных
ученых
,
вопрос
патогенеза
IgA-
нефропатии
остается
далеко
не
полностью
изученным
.
Известно
о
нарушении
процесса
полимеризации
IgAl,
образовании
антител
(IgG)
к
недостаточно
гликозилированному
IgAl,
формировании
и
выбросе
в
циркуляцию
иммунных
комплексов
,
содержащих
IgAl
с
тропностью
к
мезангиальной
депозиции
,
снижении
клиренса
IgA
и
lgA-
содержащих
иммунных
комплексов
из
системного
кровообращения
и
из
мезангия
;
афинности
мезангиальных
клеток
и
их
реакции
(
активация
)
на
мезангиальную
аккумуляцию
IgA.
Имеются
доказательства
вовлечения
ряда
генетических
факторов
.
Клиническая
картина
.
Первичная
IgA-
нефропатия
наиболее
часто
проявляется
повторными
эпизодами
макрогематурии
,
сопровождающими
респираторную
инфекцию
или
воспаление
других
слизистых
оболочек
.
В
отличие
от
ОПГН
изменения
в
анализах
мочи
появляются
уже
через
несколько
часов
от
начала
инфекции
(
рис
. 10.1.1).
В
других
случаях
может
отмечаться
микрогематурия
и
\
или
небольшая
протеинурия
,
которые
часто
обнаруживаются
при
случайном
обследовании
.
Редко
заболевание
может
проявиться
нефротическим
синдромом
и
иметь
быстропрогрессирующий
характер
.
Еще
реже
отмечается
острый
нефритический
синдром
,
напоминающий
таковой
при
ОПГН
.
У
отдельных
пациентов
в
дебюте
может
отмечаться
почечная
недостаточность
.
Гипертензия
обычно
развивается
позже
.
Уровень
фракций
комплемента
обычно
в
норме
.
Уровень
IgA
сыворотки
крови
не
имеет
решающего
диагностического
значения
:
он
бывает
повышенным
в
35—50%
случаев
.
Течение
.
IgA-
нефропатия
является
медленно
прогрессирующим
заболеванием
.
Около
25%
пациентов
нуждаются
в
гемодиализе
через
10
лет
от
начала
заболевания
.
Клиническими
факторами
риска
прогрессирования
IgA-
нефропатии
являются
гипертензия
,
протеинурии
> 1
г
/1,73
м
2,
мужской
пол
,
персистирующая
микрогематурия
.
Предикторы
прогрессирующего
течения
у
взрослых
,
такие
как
почечная
недостаточность
в
дебюте
заболевания
и
персистирующая
протеинурия
,
редко
встречаются
у
детей
.
В
то
же
время
степень
протеинурии
> 1
г
/1,73
м
2
является
известным
фактором
риска
прогрессирующего
течения
у
детей
.
В
случае
наличия
эпизодов
макрогематурии
без
протеинурии
прогноз
обычно
значительно
лучше
.
Терапия
.
С
целью
планирования
терапии
важна
идентификация
пациентов
с
факторами
риска
прогрессирования
заболевания
.
Большинство
детей
с
IgA-
нефропатией
,
в
частности
в
случае
отсутствия
или
минимального
уровня
белка
в
моче
,
не
нуждаются
в
специфической
терапии
.
Эффективность
длительного
лечения
преднизолоном
и
цитотоксическими
препаратами
при
выраженной
протеинурии
и
медленном
прогрессировании
заболевания
остается
дискутабельной
.
В
отдельных
исследованиях
рыбий
жир
(
эйкозапентаеновая
кислота
)
способен
уменьшить
протеинурию
.
Циклоспорин
неэффективен
,
в
том
числе
и
в
плане
предотвращения
рецидивов
заболевания
в
почечном
трансплантате
.
Пациенты
с
протеинурией
и
/
или
гипертензией
нуждаются
в
лечении
ингибиторами
АПФ
.
При
быстропрогрессирующем
течении
заболевания
и
наличии
полулуний
в
биоптате
проводится
терапия
преднизолоном
(
включая
пульс
-
терапию
)
и
циклофосфамидом
.
Необходимость
тонзиллэктомии
является
предметом
активных
дискуссий
в
литературе
.
Она
может
способствовать
значительному
урежению
эпизодов
макрогематурии
,
однако
роль
ее
в
отношении
длительного
прогноза
остается
неясной
.
По
-
видимому
,
она
показана
у
отдельных
больных
,
у
которых
миндалины
являются
отчетливым
очагом
рецидивирующей
инфекции
.
PDF created with pdfFactory trial version