ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 19.11.2021
Просмотров: 3129
Скачиваний: 169
эмбриональных
клеток
,
которые
потенциально
способны
перерасти
в
опухоль
Вильмса
.
Нефробластоматоэ
характеризуется
наличием
многочисленных
очагов
нефрогенных
остатков
.
Эти
изменения
могут
быть
выявлены
как
внутри
опухоли
,
так
и
в
нормальной
паренхиме
почек
у
приблизительно
1
\
3
пациентов
с
нефробластомой
.
Другие
опухоли
почек
у
детей
Светлоклеточная
саркома
—
вариант
почечной
опухоли
,
которая
может
распространиться
в
легкие
,
кости
,
головной
мозг
и
мягкие
ткани
.
Рабдоидные
опухоли
почек
отмечаются
у
детей
младше
2
лет
быстро
растут
и
распространяются
чаще
в
легкие
и
головной
мозг
.
Нейроэктодермальные
опухоли
почек
и
почечно
-
клеточная
карцинома
,
редко
встречающиеся
у
детей
,
являются
чаще
всего
заболеваниями
молодых
взрослых
.
Они
быстро
растут
и
распространяются
.
Вторичные
почечные
новообразования
могут
быть
результатом
лейкемоидной
инфильтрации
,
которая
встречается
достаточно
часто
и
приводит
к
значительному
увеличению
размеров
почек
.
Более
того
,
лимфома
может
метастазиро
вать
в
почку
или
даже
проявиться
в
виде
первичной
опухоли
почек
.
Методы
диагностики
и
определение
стадии
опухолей
Пальпация
живота
должна
проводиться
осторожно
во
избежание
разрыва
опухоли
.
В
случае
обнаружения
абдоминальных
масс
необходимо
исключить
аномалии
развития
мочеполовой
системы
.
Специфических
маркеров
опухоли
Вильмса
,
аналогичных
катехоламинам
при
нейробластоме
или
альфа
-
фетопротеину
при
гепатобластоме
,
не
существует
.
Необходимо
измерить
АД
,
провести
анализ
крови
с
учетом
клеток
и
биохимических
параметров
,
анализ
мочи
,
исследовать
свертывающую
систему
крови
.
Различные
методы
визуализации
имеют
свои
специфические
преимущества
и
направлены
на
выявление
внутрипочечных
и
внепочечных
проявлений
опухоли
,
включая
опухолевый
тромбоз
почечной
и
нижней
полой
вены
,
а
также
легочные
метастазы
.
Обычно
начинают
с
УЗИ
,
которое
в
большинстве
случаев
позволяет
определить
орган
,
из
которого
исходит
опухоль
.
За
ним
следует
компьютерная
томография
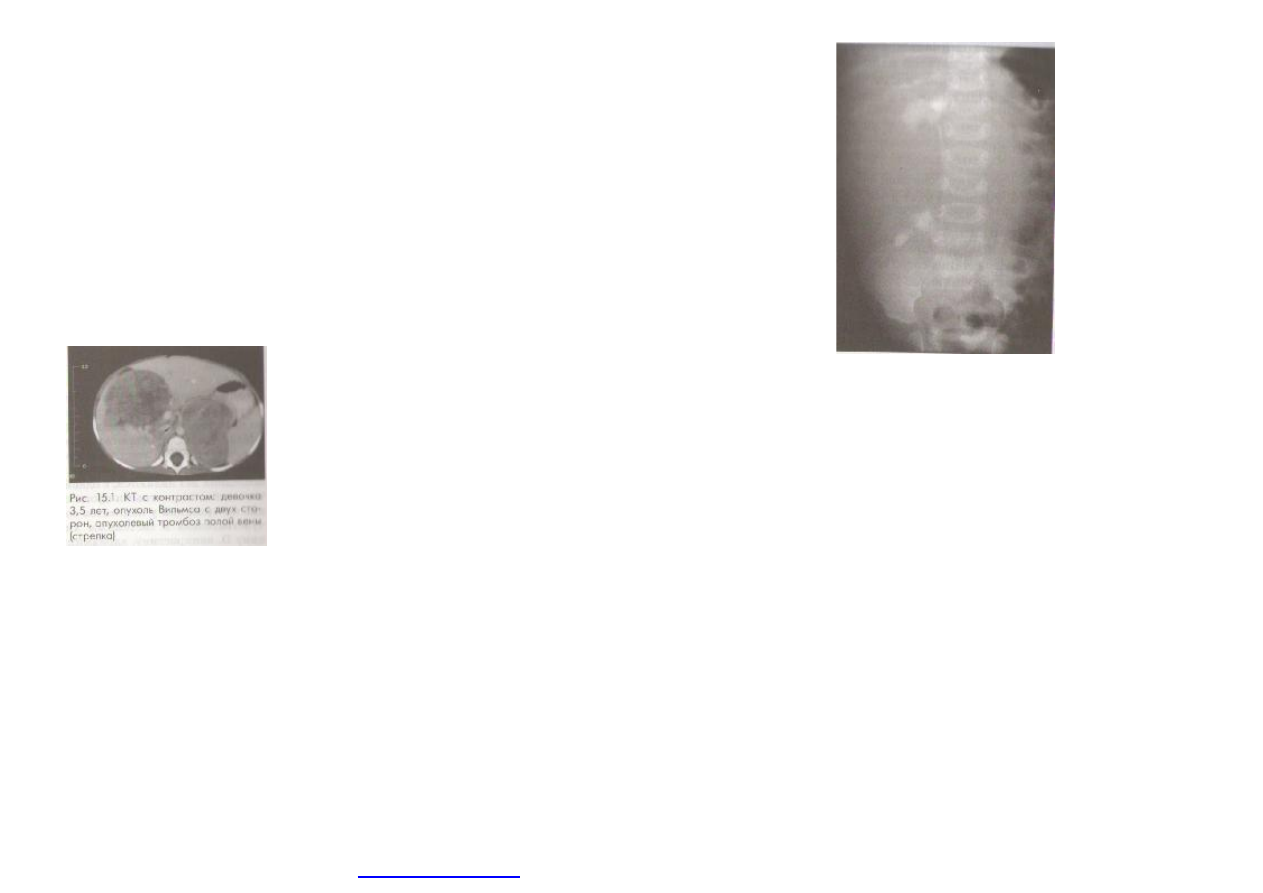
(
рис
. 15.1)
или
,
относительно
с
недавнего
времени
,
магнитно
-
резонансная
томография
(
МРТ
).
Внутривенная
урография
(
рис
.
15.2)
в
большинстве
своем
вытеснена
другими
методами
.
У
пациентов
со
светлоклеточной
саркомой
или
рабдоидной
опухолью
почки
проводят
рентгенографию
костей
и
КТ
головного
мозга
.
Определение
стадии
опухоли
проводится
с
учетом
проведенного
лечении
,
что
позволяет
планировать
дальнейшие
исследования
,
и
разработать
терапевтическую
стратегию
.
217
Рис
. 15.2.
Внутривенная
урография
:
мальчик
9
месяцев
с
большой
внутрипочечной
опухолью
Вильмса
,
переместившей
правую
лоханку
Лечение
Терапия
опухоли
почек
включает
в
себя
хирургическое
удаление
,
химиотерапию
и
лучевую
терапию
.
Кооперативные
группы
,
такие
как
Национальная
группа
по
изучению
опухоли
Вильмса
(National Wilms Tumor study group — NWTS)
и
Международное
общество
детских
онкологов
(International Society of Pediatric
Oncology — SIOP),
имеют
давнюю
традицию
организации
исследований
терапевтических
возможностей
при
этом
заболевании
.
Основными
тенденциями
последних
лет
являются
,
с
одной
стороны
,
интенсификация
терапии
у
пациентов
с
неблагоприятными
прогностическими
признаками
,
с
другой
—
уменьшение
интервенций
у
пациентов
со
стандартным
уровнем
риска
.
Нефробластома
чувствительна
к
системным
химиотерапевтическим
препаратам
:
актиномицину
D,
винкристину
,
адриамицину
,
циклофосфамиду
,
а
также
к
цисплатину
,
ифосфамиду
,
этопозиду
и
карбоплатину
.
Выбор
комбинации
препаратов
зависит
от
терапевтической
тактики
,
гистопатологии
и
стадии
заболевания
.
Основой
терапии
опухоли
Вильмса
является
хирургическое
удаление
.
Обычно
используется
трансабдоминальный
трансперитонеальный
доступ
.
В
зависимости
от
тактики
лечения
опухоль
либо
удаляется
сразу
,
либо
после
предварительной
химиотерапии
.
Группа
NWTS
рекомендует
удаление
опухоли
сразу
после
установления
диагноза
с
последующей
химиотерапией
,
которая
планируется
с
учетом
распространенности
(
стадии
)
заболевания
и
данных
гистопатологии
.
Группа
SIOP
рекомендует
предварительную
химиотерапию
.
При
этом
для
подтверждения
диагноза
возможно
проведение
игольной
биопсии
.
Последующая
химиопрофилактика
планируется
в
зависимости
от
гис
-
218
PDF created with pdfFactory trial version

топатологии
и
стадии
удаленной
опухоли
.
Лучевая
терапия
является
важным
вспомогательным
вариантом
терапии
опухоли
Вильмса
,
также
проводится
с
учетом
гистопатологии
и
стадии
заболевания
.
Прогноз
По
данным
результатов
многоцентровых
исследований
,
общая
выживпсмость
при
опухоли
Вильмса
сегодня
составляет
85%.
В
случае
установления
диагноза
на
ранних
стадиях
она
достигает
95%,
в
то
время
как
в
поздней
стадии
и
при
метастазировании
эта
цифра
снижается
до
65-70%.
Литература
1. Driscoll
К
., Isakoff
М
., Ferrer F. Update
оп
pediatric genitourinary oncology. Curr Opin
Urol 2007; 17: 281-286.
2. Rivera M.N., Haber Dji. Wilms' tumour: connecting tumorigenesis and orgatn
development in the kidney. Nat Rev Cancer 2005; 5: 699-712.
3. Sebire N.J., Vujanic G.M. Paediatric renal tumours: recent developments, n
е
w entities
and pathological features. Histopathology 2008.
4. Varan A. Wilms' tumor in children: an overview. Nephron Clin Pract 200Hi108: 83-90.
Глава
16.
Тубулопатии
16.1.
Тубулопатии
Мартин
Конрад
Врожденные
первичные
сольтеряющие
расстройства
(
синдромы
Барттера
/
Гительмана
)
Врожденные
первичные
сольтеряющие
расстройства
часто
обобщаются
под
термином
«
синдром
Барттера
».
Все
известные
варианты
заболевания
наследуются
аутосомно
-
рецессивным
путем
и
имеют
общие
клинические
характеристики
:
•
почечную
потерю
солей
;
•
гипокалиемический
метаболический
алкалоз
;
•
нормотензивный
гиперренинемический
гиперальдостеронизм
.
Идентификация
различных
канальцевых
генетических
дефектов
и
детальные
патофизиологические
исследования
позволили
создать
более
строгую
классификацию
.
С
учетом
клинической
перспективы
,
оправдано
выделение
четырех
различных
вариантов
:
1
)
антенатальный
синдром
Барттера
(
аСБ
)
является
наиболее
тяжелым
вариантом
,
ведущим
к
развитию
полигидрамниона
и
преждевременным
родам
вследствие
избыточной
продукции
мочи
in utero
.
Часто
постнатальный
период
сопровождается
угрожающими
жизни
потерями
солей
и
жидкости
,
гипокалиемическим
алкалозом
и
системными
проявлениями
в
виде
лихорадки
,
рвоты
,
диареи
и
задержки
роста
.
Выраженная
гиперкальциурия
ведет
к
формированию
нефрокальциноза
часто
в
течение
всего
нескольких
недель
.
Генерализованные
симптомы
относятся
за
счет
повышенной
системной
и
почечной
продукции
простагландинов
,
и
лечение
индометацином
,
ингибитором
синтеза
простагландинов
,
является
эффективным
терапевтическим
выбором
.
При
аСБ
функция
фуросемидчувствительного
натрий
хлоридного
котранспортера
NKCC2
в
толстом
восходящем
отделе
петли
Генле
(
ТВЧ
)
либо
непосредственно
,
либо
косвенно
нарушена
в
результате
мутаций
в
генах
NKCC2 (SLC12A1)
или
ROMK1
(KCNJ1) (
см
.
главу
2);
2)
аСБ
ассоциированный
с
нейросенсорной
тугоухостью
.
Вызван
мутацией
эссенциальной
субъединицы
(
барттин
, BSND)
почечного
хлоридного
канала
(
С
1
С
-
Ка
и
Kb).
Тугоухость
наблюдается
всегда
,
и
потери
соли
и
жидкости
даже
более
выражены
,
чем
при
дефектах
в
NKCC2
или
ROMK.
Гиперкальциурия
и
нефрокальциноз
наблюдаются
редко
.
Пациенты
с
мутацией
BSND
имеют
более
высокий
риск
развития
прогрессирующей
почечной
недостаточности
в
зависимости
от
типа
мутации
;
3)
классический
синдром
Барттера
(
кСБ
).
Клинически
пациенты
отличаются
выраженной
гипокалиемией
и
потерей
соли
,
что
приводит
к
мышечной
слабости
и
снижению
внутрисосудистого
объема
на
первом
году
жизни
с
задержкой
220
PDF created with pdfFactory trial version
роста
.
Нефрокальциноз
,
полигидрамнион
и
преждевременные
роды
при
кСБ
наблюдаются
редко
.
Молекулярным
дефектом
,
приводящим
к
данной
форме
болезни
,
является
мутация
гена
почечного
хлоридного
канала
CLCNKB;
4)
синдром
Гительмана
(
СГ
),
гипокальциурически
-
гипомагнеземический
вариант
,
является
наиболее
частым
наследственным
сольтеряющим
заболеванием
.
У
пораженных
лиц
течение
в
детском
и
подростковом
возрасте
часто
бессимптомное
.
Частыми
клиническими
симптомами
являются
транзиторнан
мышечная
слабость
,
карпопедальные
спазмы
,
эпизоды
тетанических
судорог
и
боли
в
животе
.
В
качестве
раннего
признака
СГ
может
быть
отмечена
задержка
роста
,
но
это
наблюдается
нечасто
.
СГ
обусловлен
мутациями
в
гене
тиазидчувствительного
хлоридного
котранспортера
NCCT (SLC12A2)
дистального
извитого
канальца
.
Поскольку
натрий
является
основным
определяющим
фактором
внутрисосудистого
объема
,
важно
,
чтобы
почти
все
его
профильтровавшееся
количество
была
реабсорбировано
в
почечных
канальцах
.
Поэтому
при
синдромах
Барттера
и
Гительмана
активируются
компенсаторные
механизмы
,
направленные
на
поддержание
внутрисосудистого
объема
и
АД
.
К
ним
относится
активация
ренин
-
ангиотензиновой
оси
(
гиперальдостеронизм
),
что
усиливает
дистальную
реабсорбцию
натрия
в
обмен
на
калий
и
протоны
водорода
(
Н
+).
В
зависимости
от
подтипа
болезни
,
могут
наблюдаться
различные
симптомы
(
табл
. 16.1.1).
Таблица
16.1.1.
Клинические
признаки
первичных
сольтеряющих
болезней
с
гипокалиемией
*PGE2,
простагландин
Е
2.
Специальные
исследования
(
предпочтительно
до
начала
лечения
):
•
моча
:
натрий
,
калий
,
хлорид
,
кальций
,
магний
,
креатинин
(
для
расчета
фракционной
экскреции
);
•
плазма
:
натрий
,
калий
,
хлорид
,
магний
,
кальций
,
бикарбонат
,
мочевина
,
креатинин
,
альбумин
(
как
показатели
почечной
функции
и
гидратации
);
•
УЗИ
почек
для
выявления
нефрокальциноза
;
•
генетическое
обследование
.
Дифференциальный
диагноз
•
Применение
кландестина
с
мочегонной
целью
(
анамнез
и
токсикологичес
кое
исследование
).
•
Экстраренальные
потери
соли
и
жидкости
,
например
,
при
диарее
или
муковисцидозе
.
При
диарее
концентрация
натрия
и
хлорида
в
моче
низкая
,
в
отличие
от
сольтеряющих
почечных
расстройств
.
Трудно
дифференцировать
от
хлоридной
диареи
(
псевдоБарттер
),
когда
водянистый
стул
сложно
отличить
от
мочи
.
•
Избыток
альдостерона
(
первичный
или
вторичный
):
у
больных
отмечаются
гиперволемия
и
АГ
.
•
Семейная
гипомагнеземия
с
гиперкальциурией
и
нефрокалъцинозом
(
СГГП
):
При
этом
аутосомно
-
рецессивном
заболевании
отмечаются
выраженная
гиперкальциурия
,
нефрокальциноз
,
почечные
камни
и
прогрессирующее
снижение
СКФ
.
У
больных
отмечаются
полиурия
/
полидипсия
,
рецидивирующие
инфекции
мочевыводящих
путей
или
симптоматическая
гипомагнеземия
.
Могут
присутствовать
глазные
поражения
в
виде
нистагма
,
макулярной
колобомы
,
выраженной
миопии
.
Патофизиологической
основой
являются
нарушения
парацеллюлярной
реабсорбции
магния
и
кальция
в
толстом
восходящем
отделе
петли
Генле
.
СГГН
обусловлена
мутацией
генов
протеинов
плотных
контактов
(tight
junction)
клаудина
-16
и
клаудина
-18 (
см
.
главы
2
и
21).
Лечение
антенатального
и
классического
синдрома
Барттера
Лечение
заключается
в
восполнении
потерь
жидкости
и
электролитов
с
последующим
назначением
индометацина
,
снижающего
почечную
потерю
соли
,
воды
и
калия
.
PDF created with pdfFactory trial version

Детям
в
тяжелом
состоянии
изначально
требуется
регидратация
изотоническим
раствором
хлорида
натрия
,
в
более
легких
случаях
можно
начать
непосредственно
с
назначения
хлорида
натрия
и
хлорида
калия
,
разделенных
на
3-4
приема
в
день
.
•
Обычно
бывает
трудно
адекватно
восполнить
калий
с
достижением
его
нормального
уровня
,
но
многие
пациенты
могут
относительно
безболезненно
переносить
гипокалиемию
.
В
некоторых
случаях
может
быть
получен
эффект
от
спиронолактона
,
но
следует
опасаться
,
что
он
нарушит
физиологические
компенсаторные
механизмы
и
приведет
к
выраженной
гипотензии
.
•
Индометацин
обычно
назначается
в
дозе
0,5—1
мг
/
кг
в
день
за
4
приема
с
постепенным
повышением
дозы
до
2-3
мг
/
кг
в
день
.
Он
должен
приниматься
с
пищей
или
молоком
.
Родителей
следует
предупредить
о
возможности
гастроинтестинальных
побочных
эффектов
,
вплоть
до
ульцерации
,
и
доброкачественной
внутричерепной
гипертензии
.
Хроническое
применение
индометацина
в
неонатальный
период
может
привести
к
некротизирующему
энтероколиту
.
Другие
ингибиторы
синтеза
простагландинов
,
такие
как
ибупрофен
и
ингибиторы
СОХ
-2
также
применялись
с
положительным
эффектом
.
Лечение
синдрома
Гительмана
•
Назначение
хлорида
калия
и
хлорида
или
других
солей
магния
для
контроля
электролитов
и
кислотно
-
основного
баланса
.
Это
лечение
должно
быть
пожизненным
для
предотвращения
эпизодов
тетанических
судорог
.
Больным
рекомендуется
диета
с
высоким
содержанием
соли
.
•
Спиронолактон
или
амилорид
могут
быть
использованы
для
коррекции
выраженной
гипокалиемии
,
но
могут
вызвать
тяжелую
гипотензию
.
N
В
.
При
гипокалиемических
и
гипомагнеземических
болезнях
имеется
повышенный
риск
удлинения
интервала
QT.
Поэтому
препараты
,
которые
могут
удлинять
интервал
QT (
макролиды
,
вальпроаты
и
др
.),
должны
назначаться
с
повышенной
осторожностью
,
а
лучше
избегать
их
назначения
.
Прочие
тубулярные
расстройства
Синдром
Lowe
Синдром
Lowe,
или
окулоцереброренальный
синдром
,
наследственное
заболевание
,
наследуемоеХ
-
сцепленнымрецессивным
путем
.
Ответственный
ген
OCRL-1
кодирует
фосфатидил
-
инозитол
бифосфат
5-
фосфатазу
.
Ведущими
симптомами
являются
врожденная
катаракта
,
мышечная
гипотония
,
задержка
развития
,
почечный
синдром
Фанкони
и
рахит
.
Диагностика
.
Синдром
Lowe
следует
заподозрить
у
всех
младенцев
мужского
пола
с
врожденной
катарактой
,
поскольку
типичные
дизморфические
изменения
лица
и
аминоацидурия
могут
отсутствовать
.
Следует
исследовать
хрусталики
матери
после
расширения
зрачков
,
т
.
к
.
у
матери
может
быть
субклиническая
форма
катаракты
.
Помутнение
хрусталика
у
матери
может
отсутствовать
при
мутации
de novo
и
в
редких
случаях
у
здоровых
носительниц
мутации
.
Диагноз
может
быть
точно
подтвержден
молекулярно
-
генетическим
исследованием
.
Болезнь
Дента
Болезнь
Дента
—
аутосомно
-
рецессивное
расстройство
,
вызванное
мутацией
хлоридного
канала
CLCN5,
участвующего
в
процессе
эндоцитоза
в
проксимальных
канальцах
,
что
объясняет
присутствие
тубулярной
протеинурии
у
больных
.
Недавно
у
пациентов
с
фенотипическими
проявлениями
болезни
Дента
была
обнаружена
мутация
ORCL-1,
обычно
присущая
синдрому
Lowe.
Клинические
проявления
сильно
варьируют
и
представлены
низкомолекулярной
протеинурией
,
гиперкальциурией
,
нефрокальцинозом
,
рахитом
и
ХПН
.
Псевдогипоальдостеронизм
(
ПГА
)
ПГА
представлен
гетерогенной
группой
клинических
форм
,
возникающих
вследствие
неспособности
альдостерона
осуществлять
свои
основные
физиологические
эффекты
по
обеспечению
экскреции
ионов
калия
и
водорода
.
Исходя
из
этого
для
ПГА
характерны
три
основных
признака
: 1)
гиперкалиемия
, 2)
метаболический
ацидоз
и
3)
повышенный
уровень
альдостерона
плазмы
.
Только
при
II
типе
(
синдром
Гордон
)
характерна
АГ
.
Типы
ПГА
представлены
в
табл
. 16.1.2.
Лечение
.
При
I
типе
ПГА
иногда
требуется
восполнение
хлорида
натрия
в
дозах
до
50
ммоль
/
кг
в
день
.
Адекватное
восполнение
натрия
восстанавливает
циркулирующий
объем
и
повышает
экскрецию
калия
с
мочой
.
Таблица
16.1.2.
Псевдогипоальдостеронизм
.
Типы
I—III
Тип
Наследование
Клинические
и
биохимические
признаки
Молекулярная
генетика
I
АР
*
Почечные
:
потеря
соли
,
гипонатриемия
,
гиперкалиемия
,
метаболический
ацидоз
,
повышение
АП
***
и
АРП
****,
пожизненное
назначение
соли
Легкие
:
затруднение
дыхания
,
кашель
и
одышка
.
Уровни
Na+
и
С
1~
повышены
в
поте
,
слюне
,
кале
Мутации
в
трех
генах
,
кодирующих
субъединицы
эпителиального
натриевого
канала
(ENAC)
АД
**
Почечные
:
потеря
соли
,
гипонатриемия
,
гиперкалиемия
,
метаболический
ацидоз
,
повышение
АП
и
АРП
,
восполнение
соли
.
Спонтанная
ремиссия
со
временем
Мутации
гена
,
кодирующего
минералокортикоидный
рецептор
II
АД
Синдром
Gordon:
гиперкалиемия
,
гипертензия
,
гиперхлоремический
ацидоз
,
нормальный
АП
,
низкая
АРП
,
лечение
тиазидовыми
диуретиками
По
крайней
мере
, 3
субтипа
, WNK4,
WNK1
и
др
III
Приобретенный
Гиперкалиемия
,
ацидоз
,
повышение
АП
и
АРП
,
снижение
СКФ
Вторично
к
обструктивным
уропатиям
,
серповидно
-
клеточной
и
свинцовой
нефропатии
,
амилоидозу
,
тяжелому
пиелонефриту
*
АР
—
аутосомно
-
рецессивный
, **
АД
—
аутосомно
-
доминантный
, ***
АП
—
альдостерон
плазмы
, ****
АРП
—
активность
ренина
плазмы
.
PDF created with pdfFactory trial version

Рахит
Термин
«
рахит
»
характеризует
нарушенную
минерализацию
эпифизарных
зон
роста
с
развитием
костных
деформаций
и
нарушением
линейного
роста
.
Различные
врожденные
или
приобретенные
рахитические
синдромы
могут
быть
подразделены
на
относящиеся
к
нарушениям
метаболизма
витамина
D,
гипофосфатемические
или
связанные
с
почечной
недостаточностью
.
Генерализованная
аминоацидурия
может
сопровождаться
нутритивными
(
витамин
D-
дефицитными
или
кальций
-
дефицитными
)
формами
рахита
,
что
не
свидетельствует
о
наличии
ренальных
тубулярных
расстройств
per se,
таких
как
почечный
синдром
Фанкони
.
В
табл
. 16.1.3
обобщаются
общие
клинические
и
биохимические
признаки
наследственных
рахитических
синдромов
,
включая
их
терапию
.
Таблица
16.1.3.
Наследственные
типы
рахита
.
Причины
и
биохимические
особенности
225
Нефрогенный
несахарный
диабет
Нефрогенный
несахарный
диабет
(
ННД
)
характеризуется
нечувствительносттью
канальцев
к
антидиуретическому
гормону
(
аргинин
вазопрессин
,
АВП
),
что
приводит
к
выделению
больших
количеств
разведенной
мочи
.
Выделяют
врожденную
(
тяжелую
)
и
приобретенную
(
более
легкую
)
форму
болезни
.
Наиболее
частой
генетической
формой
является
Х
-
сцепленная
с
мутацией
в
гене
,
кодирующем
рецептор
к
АВП
в
клетках
собирательных
канальцев
(V2R).
Активируясь
при
связывании
с
вазопрессином
,
рецептор
V2R
вызывает
повышение
цАМФ
,
что
в
свою
очередь
приводит
к
передвижению
в
сторону
апикальной
мембраны
внутриклеточных
везикул
,
содержащих
водные
каналы
аквапорина
-2 (AQ-2)
повышает
проницаемость
канальцев
для
воды
.
Некоторые
мутации
прерывают
функцию
рецепторов
полностью
,
тогда
как
другие
лишь
снижают
аффинность
рецепторов
к
АВП
,
что
сопровождается
более
легким
фенотипическими
проявлениями
(
частичный
ННД
).
Женщины
—
носительницы
мутации
V2R
в
семьях
с
ННД
обычно
бессимптомны
.
Редко
женщины
из
семей
с
Х
-
сцепленным
ННД
имеют
инактивированную
вторую
Х
-
хромосому
,
что
приводит
к
псевдодоминантному
наследованию
болезни
.
В
редких
случаях
НДД
может
наследоваться
по
аутосомно
-
рецессивному
пути
.
Пораженные
лица
имеют
мутации
в
гене
,
кодирующем
протеин
аквопорина
-2.
При
полном
и
не
леченном
ННД
осмоляльность
мочи
обычно
находится
в
пределах
50—100
мОсм
/
кг
.
Общий
объем
экскретируемой
мочи
зависит
от
осмотической
нагрузки
(
осмотически
активных
веществ
в
питании
),
которая
должна
быть
экскретирована
почками
.
С
обычной
диетой
нагрузка
на
почки
составляет
около
500
мОсм
в
день
.
При
осмоляльности
мочи
50
мОсм
/
кг
для
экскреции
осмотической
нагрузки
потребуется
10
л
воды
.
Лишний
грамм
соли
(=
по
18
ммоль
натрия
и
хлорида
каждого
=36
мОсм
)
в
диете
приведет
к
увеличению
продукции
мочи
примерно
на
700
мл
.
226
PDF created with pdfFactory trial version