ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 30.11.2023
Просмотров: 154
Скачиваний: 2
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.
2)Пайда болу себебі: Бұл аурудың генін «Rb гені» деп атайды. Бұл қатерлі ісік дамуына жауап беретін алғашқы табылған және көп зерттелген гендердің бірі. Оның сипаттамалары:13q14.1 хромосоманың ұзын қолының ортаңғы бөлімінде орналасқан;27 экзоннан тұрады;геномдық ДНҚ-ның 180 000 базалық жұбын алады;қалыпты жағдайда бүкіл ағзаның жасушаларында көрінеді. 50-60% жағдайда ретинобластома генетикалық (туа біткен) ауру. Ретинобластомамен ауыратын балалардың жартысынан көбі балалық шағында осындай ауруға шалдыққан ата-аналардан туады. Ретинобластоманың спорадикалық жағдайлары сирек кездеседі және белгісіз себептермен пайда болады. Ретинобластоманың генетикалық емес формасы үшін қауіп факторларының қатарына ата-аналардың жоғары жасын, олардың металлургиялық өндіріс орындарында жұмыс жасауын, торлы қабық пен жыныс жасушаларының хромосомаларында ауытқулар тудыратын қолайсыз экологиялық және тамақтану факторларын қосу жатқызылады.
3)Зерттеу әдістері: анамнезді жинау;балалардың шағымдарын бағалау;физикалық тексеру;аспаптық зертттеулер;дәрігер тұқым қуалайтын фактордың болуына ерекше назар аударады,офтальмологиялық тексеру,көзді УДЗ тексеру,бас миыныа және орбитаға МРТ жасау, биомикроскопия, тура және кері офтальмоскопия, висометрия, тонометрия, гониоскопия, бинокулярлық көру, экзофтальмометрия, страбизм бұрышын өлшеу.
Муковицидоз- аутосомды-рецессивті ауру,тіндердің зақымдануымен және экзокринді бездердің секреторлық қызметінің бұзылуымен, сондай-ақ функционалдық бұзылыстармен, ең алдымен тыныс алу және ас қорыту жүйелерінің бұзылыстарынан көрінетін,туа біткен ауыр ауру.
1)Пайда болу себебі: үш негізгі пункт жетекші болып табылады: сыртқы секреция бездерінің зақымдануы, дәнекер тіннің өзгеруі, су-электролит бұзылыстары.Муковицидоздың себебі ген мутациясы болып табылады, су-электролиттік метаболизміне қатысатын CFTR ақуызының (цистикалық фиброздың трансмембраналық реттегіші) құрылымы мен қызметі бұзылады нәтижесінде бронхопульмониялық жүйені, ұйқы безін, бауырды, асқазан-ішек жолдарын және репродуктивті органдарды жабатын эпителийдің құрылымы мен қызметі бұзылады.
2)Клиникалық көріністері: Цистозды фиброз кезінде аурудың белгілері көбінесе бала туылғаннан кейін алғашқы айларда көрінеді және ішек өтімсіздігінің белгілерімен көрінеді (меконий илеус):қатты мазасыздық және үнемі жылау;іштің кебулері;құсу;сілекейдің жоғарылауы;нәжісті ұзақ уақыт ұстау;дене температурасының жоғарылауы;жиі диарея;майлы тағамға төзбеушілік;тән нәжіс - тұтқыр, жабысқақ, майлы.Кейінірек балада ақыл-ой кемістігі,физикалық тұрғыдан артта қалу,тершеңдік,жыныстық жетілуі кешеуілдеу.
3)Диагностикалау әдістері:
-
Отбасылық және тұқым қуалаушылық тарихын, аурудың алғашқы белгілерін, клиникалық көріністерін зерттеу; -
Қан мен зәрді жалпы талдау; -
Копрограмма - майдың, талшықтың, бұлшықет талшықтарының, крахмалдың болуы мен құрамына арналған нәжісті зерттеу (ас қорыту жолдары бездерінің ферментативті бұзылыстарының дәрежесін анықтайды); -
Қақырықты микробиологиялық зерттеу; -
Бронхография (бронхэктазаның, «тамшы тәрізді» тән бронхиалды ақаулардың болуын анықтайды) -
Бронхоскопия (бронхтарда жіптер түрінде қалың және тұтқыр қақырықты анықтайды); -
Өкпенің рентгенографиясы (бронхтар мен өкпелердегі инфильтративті және склероздық өзгерістерді анықтайды); -
Спирометрия (дем шығарған ауаның көлемі мен жылдамдығын өлшеу арқылы өкпенің функционалдық күйін анықтайды); -
Пот тесті - тер электролиттерін зерттеу - муковисцидозға арналған негізгі және ақпаратты талдау (муковисцидозы бар науқастың терінде хлор мен натрий иондарының көп мөлшерін анықтауға мүмкіндік береді); -
Молекулалық-генетикалық тестілеу (қандағы немесе ДНҚ сынамаларына муковисцидоз генінде мутацияның бар-жоғын анықтау);
Пренатальды диагноз - жаңа туған нәрестелерді генетикалық және туа біткен ауруларға тексеру.
Тея-Сакса ауруы – Ми мен жұлынның нейрондарында липоидты макромолекулалар жиналуымен,гексоаминизаза фермент өндірісінің және бетаНацетилгексозаминидаза фермнің альфа суббнің кодталуына жауап беретін метаболиттік бұзылуымен,аутосомдырецессивті типпен тұқым қуалайтын,әрі ол 15хромосоманың ұзын иығының 2аудан 3сегментінде орналасқан HEXA генінің аберрациясынан туындайды. Клиникалық белгілері науқастың шалдыққан жасына байланысты сипатталады. Нәресте туылғаннан кейінгі алғашқы алты айда қалыпты дамиды. Содан кейін, нейрондарды GM2 ганглиозидтері созған кезде, ақыл-ой мен физикалық қабілеттің шексіз төмендеуі басталады. Бала соқыр, саңырау,ақыл-ой қабілетінің прогрессивті жоғалуы,деменция,шуылдың жоғарылауы,прогрессивті есту қабілетінің жоғалуы,жұтылу қиындықтары, баланың екінші жылында басталуы мүмкін ұстамалар және олардың көздеріндегі шие-қызыл дақтар, атрофияланған және параличке айналады. Өлім әдетте төрт жасқа дейін болады.
Кәмелетке толмаған кезінде ауру екі жастан он жасқа дейінгі балаларда байқалады. Бұл типте когнитивті және моториканың, дизартрияның, дисфагияның, атаксияның және спастиканың нашарлауына ұшырайды. Өлім бес пен он бес жас аралығында болады.
Кеш бастау сирек кездесетін түрі ересектерде, әдетте 20-40 жас аралығында алғашқы белгілері көрінеді. Осы түрінің айырмашылығы, өлімге әкелмейді, өйткені оның әсері ілгерілеуді тоқтатуы мүмкін. Бұл жиі дұрыс диагноз қойылмайды. Бұл тұрақсыз жүріспен және үдемелі неврологиялық нашарлаумен сипатталады. Белгілеріне сөйлеу мен жұтынудың қиындауы , жүрістің тұрақсыздығы, спастикалық, когнитивті құлдырау және психикалық аурулар, әсіресе психоз жатады.шизофренияға ұқсас. Ересек жаста мүгедектер арбасының тұрақты пайдаланушысы болады.
Диагностикасы: Биохимиялық қан сынамасы не ферменттік талдауға негізделеді. Ферменттерді талдау - бұл адамның қанындағы деңгейін гексоаминизаза өлшейтін биохимиялық тест.
ДНҚ тасымалдаушының сынағы- HexA кодтайтын геннің белгілі бір мутациясын немесе өзгеруін іздейді.
Пренатальды тестілеу- жүктіліктің 11-ші аптасында хорионды вилли сынамаларын (CVS) қолдану арқылы жүргізуге болады. Бұл плацентаның кішкене бөлігін алып тастаудан тұрады. Сонымен қатар, ұрықты жүктіліктің 16-шы аптасында амниоцентезбен тексеруге болады. Бұл процедурада ине нәрестені қоршаған сұйықтықтың сынамасын алу және тексеру үшін қолданылады.
Офтальмофтың көмегімен көз қарашығының қызыл дағын анықтау,тері биопсиясы, нейрондарды микроскопиялық зерттеу ұсынылады.
Емдеуі: Қазіргі уақытта Тай-Сакс ауруын емдеудің нақты әдісі жоқ. Антиконвульсандық дәрі-дәрмектер ұстаманы басқара алады. Қолдаушы басқа емдеу әдісі дұрыс тамақтану мен ылғалдандыруды және тыныс алу жолын ашық ұстау әдістерін қамтиды. Балаларға ақыр соңында тамақ беретін түтік қажет болуы мүмкін
Ниманн-Пик ауруы-бұл липидтер алмасуының бұзылуынан сфингомиелиннің, бауыр, көкбауыр,өкпе, сүйек кемігі мен ми жасушаларының лизосомаларында жинақталуынан туындайтын аутосомды-рецессивті түрде тұқым қуалайтын ауру. Аурудың үш түрі бар: А, В және С типтері .
Аурудың диагнозы: Фенилкетонурия
Аурудың тұқым қуалау түрі:аутосомды рецессивті
Жиілік: 1:10 000
Фенилкетонурия- фенилаланин метаболизмінің бұзылуына байланысты пайда болатын ауру,яғни финилаланиннің тирозин аминқышқылына айналмауы. Көбінесе, фенилкетонурияның дамуы фенилаланин-4-гидроксилаза ферментін кодтайтын геннің мутациясы салдарынан болады, ол 12 хромосоманың ұзын иығында орналасқан (локус 12q22-q24.1). Бала туылғанда сау болып туылады.Аурудың симтомдары үш-алты айдан кейін көріне бастайды, яғни фенилаланинді тамақпен қабылдау өмірдің бірінші жартысында аурудың көрінісін туғызады, содан кейін баланың дамуында ауыр бұзылуларға әкеледі.Фенотиптік көріністері:ақшыл тері,шашының ақшыл болуы,көзінің көк болуы байқалады.
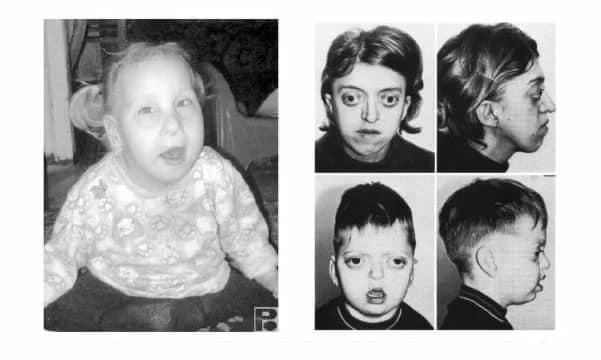
Клиникалық көріністеріне тоқталатын болсам:
-
Неврологиялық және психикалық бұзылулар; -
Ақыл-ойдың артта қалуы; -
Балалық шақта мазасыздықтың жоғарылауы; -
Ерекше жүріс; -
Ерекше қалып және ерекше отырыс қалпы; -
Аяқтың әдеттен тыс орналасуы; -
Тері өзгерістері; -
Ұстамалар; -
Экзема; -
Жаңа туған кезеңдегі құсу; -
Зәрінен тышқанның иісі шығады,себебі фенилаланин болады құрамында зәрдің.
Әр түрлі тұқым қуалайтын метаболикалық ақаулар әр түрлі жиілікте пайда болады. Фенилкетонурия көбінесе анықталады - 6000 баланың 1-інде - 10000 жаңа туылған нәрестелер: бұл ақыл-есі кем науқастардың контингенті арасында шамамен 1% құрайды
Диагностика әдістері бізде:
ПКУ негізгі биохимиялық маркері - фенилаланиннің плазмалық концентрациясы ны ң жоғарылауы ( гиперфенилаланинемия ) - тамақтану басталғаннан 3-4 күн өткен соң анықталады
Биохимиялық диагностикалық критерийлер:
- Феллинг тесті (скринингтік тест): жасыл зәр;
- биофан Р көмегімен индикаторлық қағаз сынағы ;
- Гутри тесті ;
- « Флюроскопты » қолданатын иммуно- ферменттік әдіс .
- плазмадағы фенилаланин мөлшері 200 мг / л жоғары;
- тирозиннің плазмадағы қалыпты деңгейі;
- зәрдегі фенилаланин метаболиттерінің ( фенилпировтық және гидроксифенил қышқылдар ы) деңгейінің жоғарылауы , тышқанның зәрінің иісі;
- іштен алынған фенилаланинге төзімділіктің төмендеуі ;
- тетрагидробиоптерин кофакторының қалыпты төзімділігі .
OMIM базасы бойынша осы аурудың дамуына жауапты геннің локализациясы: (PKU) (E70.1) 12q24.1 RAS, PKU1 немесе - дигидроптеридин- редуктаза тапшылығы жағдайында - 4q15.1
Осы аурудың клиникалық белгісін жүргізудің молекулалық-генетикалық механизмдерін түсіндіріңіз:
Фенилкетонурия (фенилаланинкетонурия, фенилпирожүзімдік олигофрения, ФКУ) - фенилаланингидроксилаза(ФАГ) ферментінің белсенділігінің жетіспеушілігіне (болмауы не төмендеуіне) байланысты ауру. Метаболизмдік блоктың нәтижесінде фенилаланин метаболизмінің жанама жолдары активтенеді, ал организмде оның іс жүзінде қалыпты қалыптаспаған улы туындылары - фенилпирувий және фенил сүт қышқылдары жинақталады. Сонымен қатар, фенилэтиламин және ортофенил ацетат, олар нормада мүлдем жоқ болуы керек, сонымен қатар олардың артық мөлшері мидағы липидтер алмасуының бұзылуын тудырады. Болжам бойынша, мұндай науқастарда ақылдылықтың ақымақтық деңгейіне дейін прогрессивті төмендеуіне әкеледі. Ақырында, фенилкетонурия кезінде ми дисфункцияларының даму механизмі түсініксіз болып қалады. Себептер қатарында ми-нейротрансмиттердің жетіспеушілігі тирозин мен гематоэнцефалдық бөгет арқылы тасымалданғанда фенилаланинмен бәсекелес басқа «ірі» аминқышқылдарының салыстырмалы түрде азаюынан және фенилаланиннің тікелей әсерінен туындайды.Фенилаланиннің соңғы өнімдері меланиннің жеткіліксіздігін тудырады.Сол себепті клиникалық белгілерінде ақ шаш және көк көзділік байқалады.
Емдеу жолдарына келетін болсам:
Емдеу ауруды анықтағаннан бастап жыныстық жетілуге дейін қатаң диета түрінде жүзеге асырылады, көптеген дәрігерлер өмір бойы тамақтану режимі керек деген пікірде. Диетада ет, балық, сүт өнімдері және жануарлардан және ішінара өсімдік ақуызынан тұратын басқа өнімдер жоқ тағамдармен тамақтануы қажет. Ақуыздың жетіспеушілігі фенилаланинсіз аминқышқыл қоспаларымен толтырылады. Фенилкетонуриямен ауыратын балаларды емізу мүмкін және кейбір шектеулер сақталған жағдайда сәтті болады
Гемофилия - бұл қан кетудің жоғарылауымен сипатталатын тұқым қуалайтын ауру.Тұқым қуалау типі-Х тіркес ресессивті,көбіне ер адамдар ауырады,әйелдер тасымалдаушы болып табылады.