ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 23.08.2024
Просмотров: 2214
Скачиваний: 1
СОДЕРЖАНИЕ
Основные понятия и законы химии Атомно - молекулярное учение.
Закон сохранения массы веществ
Составление химических уравнений
Расчеты по химическим уравнениям
Закон Авогадро и молярный объем газа
Основные классы неорганических
2. Разложение некоторых кислородсодержащих веществ (оснований, кислот, солей) при нагревании:
Графическое изображение формул
Химическое равновесие. Константа химического равновесия
Энергия Гиббса направленность химических процессов
Примеры термодинамических расчетов
Химическая кинетика. Скорость химической реакции
Молекулярность элементарных реакций
Смещение химического равновесия
3 Влияние температуры на положение равновесия
Основные характеристики растворов
Растворимость газов в жидкостях
Взаимная растворимость жидкостей
Растворимость твердых веществ в жидкостях
1. Давление насыщенного пара разбавленных растворов
2. Давление пара идеальных и реальных растворов
3. Температура кристаллизации разбавленных растворов
4. Температура кипения разбавленных растворов
5. Осмотическое давление разбавленных растворов
6. Понятие активности растворенного вещества
Слабые электролиты. Константа диссоциации
Количественные характеристики процесса гидролиза соли.
Направленность реакций в растворах электролитов
Протонная теория Брёнстеда-Лоури
Коррозия металлов и методы защиты металлов от коррозии
Волновое уравнение. Квантовомеханическое объяснение строения атома
Электронная структура атомов и периодическая система элементов
Структура периодической системы элементов д.И. Менделеева.
Периодичность свойств химических элементов и их соединений
Ковалентная связь. Метод валентных связей
Способы образования ковалентной связи
Гибридизация атомных орбиталей
Квантовомеханические теории строения комплексных соединений
2. Гибридизация орбиталей и структура комплексов
Единицей измерения скорости является [моль/(дм3 с)]. В различных интервалах времени средняя скорость химической реакции имеет разные значения; истинная (мгновенная) скорость реакции определяется как производная от концентрации по времени:
![]()
Графическое изображение зависимости концентрации реагентов от времени есть кинетическая кривая (рисунок 1):
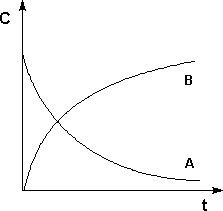
Рис. 1. Кинетические кривые для исходных веществ (А) и продуктов реакции (В).
Истинную скорость реакции можно определить графически, проведя касательную к кинетической кривой; истинная скорость реакции в данный момент времени равна по абсолютной величине тангенсу угла наклона касательной:
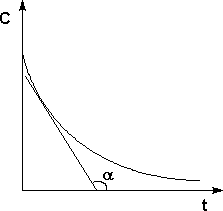
Рис. 2Графическое определение Vист.
![]()
Необходимо отметить, что в том случае, если стехиометрические коэффициенты в уравнении химической реакции неодинаковы, величина скорости реакции будет зависеть от того, изменение концентрации какого реагента определялось. Очевидно, что в реакции
2Н2 + О2 ––> 2Н2О
концентрации водорода, кислорода и воды изменяются в различной степени: ΔС(Н2) = ΔС(Н2О) = 2 ΔС(О2).
Молекулярность элементарных реакций
Элементарными (простыми) называют реакции, идущие в одну стадию. Их принято классифицировать по молекулярности:
Молекулярность элементарной реакции – число частиц, которые, согласно экспериментально установленному механизму реакции, участвуют в элементарном акте химического взаимодействия.
Мономолекулярные – реакции, в которых происходит химическое превращение одной молекулы (изомеризация, диссоциация и т. д.):
I2 ––> I• + I•
Бимолекулярные – реакции, элементарный акт которых осуществляется при столкновении двух частиц (одинаковых или различных):
СН3Вr + КОН ––> СН3ОН + КВr
Тримолекулярные – реакции, элементарный акт которых осуществляется при столкновении трех частиц:
О2 + NО + NО ––> 2NО2
Реакции с молекулярностью более трёх неизвестны.
Одной из задач, стоящих перед химической кинетикой, является определение состава реакционной смеси (т.е. концентраций всех реагентов) в любой момент времени, для чего необходимо знать зависимость скорости реакции от концентраций. В общем случае, чем больше концентрации реагирующих веществ, тем больше скорость химической реакции. В основе химической кинетики лежит т. н. основной постулат химической кинетики:
Скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в некоторых степенях.
Для реакции
аА + bВ + dD + ... ––> еЕ + ...
можно записать:
![]()
Коэффициент пропорциональности k есть константа скорости химической реакции. Константа скорости численно равна скорости реакции при концентрациях всех реагирующих веществ, равных 1 моль/л.
Зависимость скорости реакции от концентраций реагирующих веществ определяется экспериментально и называется кинетическим уравнением химической реакции. Очевидно, что для того, чтобы записать кинетическое уравнение, необходимо экспериментально определить величину константы скорости и показателей степени при концентрациях реагирующих веществ. Показатель степени при концентрации каждого из реагирующих веществ в кинетическом уравнении химической реакции (в уравнении соответственно x, y и z) есть частный порядок реакции по данному компоненту. Сумма показателей степени в кинетическом уравнении химической реакции (x + y + z) представляет собой общий порядок реакции. Следует подчеркнуть, что порядок реакции определяется только из экспериментальных данных и не связан со стехиометрическими коэффициентами при реагентах в уравнении реакции. Стехиометрическое уравнение реакции представляет собой уравнение материального баланса и никоим образом не может определять характера протекания этой реакции во времени. Для элементарных реакций, проводимых при близких концентрациях исходных веществ, величины молекулярности и порядка реакции совпадают. Тем не менее, никакой чётко определенной взаимосвязи между понятиями молекулярности и порядка реакции не существует, поскольку порядок реакции характеризует кинетическое уравнение реакции, а молекулярность – механизм реакции.
Зависимость скорости химической реакции от температуры
Скорость химической реакции значительно зависит от температуры. Не всякое столкновение реагирующих частиц приводит к их взаимодействию. В химическое взаимодействие вступают только активные молекулы, т. е. обладающие энергией, достаточной для осуществления данной реакции. При повышении температуры число активных молекул возрастает, так как нагревание сообщает молекулам необходимую энергию активации, т. е. ту дополнительную энергию, которая приводит к ослаблению химических связей в молекулах реагирующих веществ, а затем и к их разрыву.
Зависимость скорости химической реакции от температуры определяется эмпирическим правилом Вант-Гоффа: при повышении температуры на каждые 10о скорость реакции возрастает примерно в 2–-4 раза.
Vt = Vt
= Vt
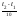 ,
,
где
Vt ,
Vt
,
Vt – скорости реакции при температурах
t2 и t1,
– температурный коэффициент скорости
реакции, который представляет собой
отношение констант скорости реакций
при температурах, отличающихся на 10
градусов
= k2 /k1 и приблизительно равен 2-4.
– скорости реакции при температурах
t2 и t1,
– температурный коэффициент скорости
реакции, который представляет собой
отношение констант скорости реакций
при температурах, отличающихся на 10
градусов
= k2 /k1 и приблизительно равен 2-4.
Такое существенное влияние температуры на скорость химической реакции нельзя объяснить только лишь увеличением числа столкновений молекул реагентов. Так как согласно молекулярно-кинетической теории газов средняя скорость молекул пропорциональна квадратному корню из абсолютной температуры, т.е. при повышении температуры, например от 300 до 310 К, средняя скорость молекул возрастет лишь в √310/300 = 1,02 – гораздо меньше, чем требует правило Вант-Гоффа.
Объяснить существенное влияние температуры на скорость химической реакции можно используя теорию активации Аррениуса.
Уравнение Аррениуса
Очевидно, что взаимодействие частиц осуществляется при их столкновениях; однако число столкновений молекул очень велико и, если бы каждое столкновение приводило к химическому взаимодействию частиц, все реакции протекали бы практически мгновенно. С. Аррениуспостулировал, что столкновения молекул будут эффективны (т.е. будут приводить к реакции) только в том случае, если сталкивающиеся молекулы обладают некоторым запасом энергии – энергией активации.
Энергия активации есть минимальная энергия, которой должны обладать молекулы, чтобы их столкновение могло привести к химическому взаимодействию.
Опытным путем установлены примерные границы энергий активации для реакций, идущих с соизмеримыми скоростями: если Еа< 50 кДж, то реакция идет с неизмеримо большой скоростью, если же Еа > 100 кДж, то скорость реакции неизмеримо мала.
Рассмотрим путь некоторой элементарной реакции
А + В ––> С
Поскольку химическое взаимодействие частиц связано с разрывом старых химических связей и образованием новых, считается, что всякая элементарная реакция проходит через образование некоторого неустойчивого промежуточного соединения (переходного состояния), называемого активированным комплексом А…В:
А ––> А…В ––> B
Образование активированного комплекса всегда требует затраты некоторого количества энергии, что вызвано, во-первых, отталкиванием электронных оболочек и атомных ядер при сближении частиц и, во-вторых, необходимостью построения определенной пространственной конфигурации атомов в активированном комплексе и перераспределения электронной плотности. Таким образом, по пути из начального состояния в конечное система должна преодолеть своего рода энергетический барьер. Энергия активации реакции приближённо равна превышению средней энергии активированного комплекса над средним уровнем энергии реагентов. Очевидно, что если прямая реакция является экзотермической, то энергия активации обратной реакции Е'А выше, чем энергия активации прямой реакции EA. Энергии активации прямой и обратной реакции связаны друг с другом через изменение внутренней энергии в ходе реакции. Вышесказанное можно проиллюстрировать с помощью энергетической диаграммы химической реакции:
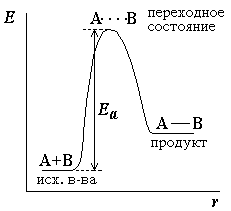
Рис. 1. Энергетическая диаграмма для эндотермической реакции образования продукта АВ из исходных веществ А и В.
Если энергия столкновения молекул А и В больше или равна энергии активации Еа, то энергетический барьер преодолевается, и происходит перемещение вдоль координаты реакцииr от исходных веществ к продукту. Иначе имеет место упругое столкновение молекул А и В. Вершина энергетического барьера соответствуетпереходному состоянию(активированному комплексу), в котором связь А–В образовалась частично.
Поскольку температура есть мера средней кинетической энергии частиц, повышение температуры приводит к увеличению доли частиц, энергия которых равна или больше энергии активации, что приводит к увеличению константы скорости реакции (рис.2.):
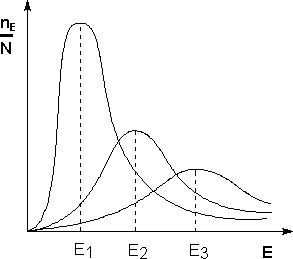
Рис. 2.Распределение частиц по энергии Здесь nЕ/N – доля частиц, обладающих энергией E; Ei- средняя энергия частиц при температуре Ti(T1< T2< T3)
Зависимость константы скорости реакции от температуры и величины энергии активации описывается – уравнением Аррениуса:
k= A e-Ea / RT,
где А – предэкспоненциальный множитель, не зависящий от температурыиконцентрации и равный числу «удачных» столкновений в единицу времени (т.е. молекулы в момент столкновения должны ориентированы друг относительно друга подходящим образом). Физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения, логарифм константы скорости линейно зависит от обратной температуры: Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
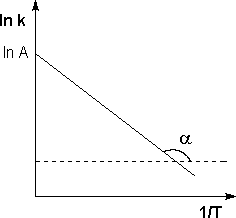
Рис. Зависимость логарифма константы скорости химической реакции от обратной температуры.
Величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
![]()
При известных значениях константы скорости реакции при разных температурах: