ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 23.08.2024
Просмотров: 2247
Скачиваний: 1
СОДЕРЖАНИЕ
Основные понятия и законы химии Атомно - молекулярное учение.
Закон сохранения массы веществ
Составление химических уравнений
Расчеты по химическим уравнениям
Закон Авогадро и молярный объем газа
Основные классы неорганических
2. Разложение некоторых кислородсодержащих веществ (оснований, кислот, солей) при нагревании:
Графическое изображение формул
Химическое равновесие. Константа химического равновесия
Энергия Гиббса направленность химических процессов
Примеры термодинамических расчетов
Химическая кинетика. Скорость химической реакции
Молекулярность элементарных реакций
Смещение химического равновесия
3 Влияние температуры на положение равновесия
Основные характеристики растворов
Растворимость газов в жидкостях
Взаимная растворимость жидкостей
Растворимость твердых веществ в жидкостях
1. Давление насыщенного пара разбавленных растворов
2. Давление пара идеальных и реальных растворов
3. Температура кристаллизации разбавленных растворов
4. Температура кипения разбавленных растворов
5. Осмотическое давление разбавленных растворов
6. Понятие активности растворенного вещества
Слабые электролиты. Константа диссоциации
Количественные характеристики процесса гидролиза соли.
Направленность реакций в растворах электролитов
Протонная теория Брёнстеда-Лоури
Коррозия металлов и методы защиты металлов от коррозии
Волновое уравнение. Квантовомеханическое объяснение строения атома
Электронная структура атомов и периодическая система элементов
Структура периодической системы элементов д.И. Менделеева.
Периодичность свойств химических элементов и их соединений
Ковалентная связь. Метод валентных связей
Способы образования ковалентной связи
Гибридизация атомных орбиталей
Квантовомеханические теории строения комплексных соединений
2. Гибридизация орбиталей и структура комплексов
Поскольку S0 незначительно зависит от температуры, то
Т
S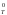
Т
S
Т
S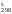 .
.
Энергия Гиббса направленность химических процессов
Для учета одновременного влияния энтальпийного (Н) и энтропийного (S) факторов при постоянных давлении и температуре используют так называемую энергию Гиббса (свободную энергию, uзобарно-uзоmермический потенциал):
G = Н – Т S,
или для стандартных условий:
G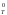 = Н
= Н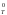 – Т
S
– Т
S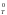 , (1)
, (1)
где
G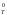 – изменение стандартной энергии Гиббса
при температуре Т.
– изменение стандартной энергии Гиббса
при температуре Т.
Расчет изменения стандартной энергии Гиббса обычно проводят по приближенному уравнению:
G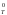
Н
Н – ТS
– ТS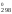 . (2)
. (2)
Обозначения:
G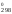 – изменение стандартной энергии Гиббса
в реакции;
– изменение стандартной энергии Гиббса
в реакции;
G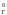 – изменение стандартной энергии Гиббса
в реакции при температуре Т;
– изменение стандартной энергии Гиббса
в реакции при температуре Т;
G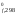 – стандартная энергия Гиббса образования
веществ и ионов из простых веществ. G
– стандартная энергия Гиббса образования
веществ и ионов из простых веществ. G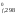 – изменение энергии Гиббса реакции
образования 1 моля вещества из простых
веществ при условии, что все участники
реакции находятся в стандартных
состояниях.
– изменение энергии Гиббса реакции
образования 1 моля вещества из простых
веществ при условии, что все участники
реакции находятся в стандартных
состояниях.
Единицы
измерения G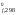 – кДж/моль; G
– кДж/моль; G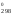 – кДж.
– кДж.
Для
расчета G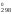 для процесса необходимо:
для процесса необходимо:
записать соответствующий процесс, указав агрегатные состояния веществ, участвующих в реакции;
расставить стехиометрические коэффициенты;
выписать из справочника величины стандартных теплот образования и стандартных энтропий всех участвующих в реакции веществ в соответствующих агрегатных состояниях;
рассчитать значения Н
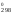 и S
и S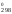 реакций, как указано выше, и, подставив
их в уравнение G
реакций, как указано выше, и, подставив
их в уравнение G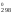 = Н
= Н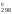 – 298·S
– 298·S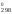 ,
найти значение G
,
найти значение G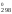 .
Как и любая термодинамическая функция,
энергия Гиббса является функцией
состояния, т.е. ее значение не зависит
от пути протекания процесса, а лишь от
исходного и конечного состояний системы.
Поэтому энергию
Гиббса химической реакции можно
рассчитать как сумму энергий Гиббса
образования продуктов реакции за
вычетом суммы энергий Гиббса образования
исходных веществ с учетом стехиометрических
коэффициентов.
Так как в таблицах приведены значения
G
.
Как и любая термодинамическая функция,
энергия Гиббса является функцией
состояния, т.е. ее значение не зависит
от пути протекания процесса, а лишь от
исходного и конечного состояний системы.
Поэтому энергию
Гиббса химической реакции можно
рассчитать как сумму энергий Гиббса
образования продуктов реакции за
вычетом суммы энергий Гиббса образования
исходных веществ с учетом стехиометрических
коэффициентов.
Так как в таблицах приведены значения
G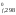 ,
то использовать такой расчет можно
лишь для G
,
то использовать такой расчет можно
лишь для G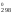 .
.
При расчете G
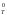 пренебрегают зависимостью Н0
и S0
от температуры и рассчитывают G
пренебрегают зависимостью Н0
и S0
от температуры и рассчитывают G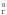 по приближенному уравнению
по приближенному уравнению
G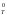
Н
Н – ТS
– ТS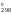 ..(2).
..(2).
По величине изменения энергии Гиббса в реакции можно судить о принципиальной термодинамической возможности осуществления процесса.
Термодинамика утверждает, что в любой закрытой системе при постоянном давлении и температуре возможен только такой самопроизвольный процесс, который ведет к уменьшению энергии Гиббса (G <0). При значениях G >0 процесс термодинамически невозможен. При G = 0 система находится в состоянии равновенсия.
Если
условия протекания процесса отличаются
от стандартных, то используют величину
G.
Значений
G
для процесса может быть получено
множество в отличие от единственного
значения G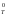 Связь между G
и
Gо
выражается уравнением, получившим
название изотермы
Вант-Гоффа
( в честь голландского физико-химика),
которая для реакции:
Связь между G
и
Gо
выражается уравнением, получившим
название изотермы
Вант-Гоффа
( в честь голландского физико-химика),
которая для реакции:
а А + в В = с С + d D
записывается в виде:
G = Gо + RT ln (PAa PBb/ PCc PDd)
или
G = Gо + RT ln (CAa CBb/ CCc CDd)
Где Р – относительные парциальные давления соответствующих веществ; С концентрации соответствующих растворенных веществ.
Если концентрации либо парциальные давления веществ, участвующих в реакции = 1, то G = Gо.
При равновесии G = 0, а выражение под логарифмом преобразуется в Кр (Кс для растворов). Отсюда получаем одно из важнейших уравнений термодинамики, связывающее константу химического равновесия с изменением стандартной энергии Гиббса:
G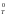 = - RTlnКр,T
= - RTlnКр,T
Если в уравнение подставить значение постоянной R = 8,314 Дж/(моль К) и ввести коэффициент перехода от натурального к десятичному логарифму 2,303, то выражение можно записать следующим образом:
G = –RT
2,303lgKp
= –19,15TlgKp.
= –RT
2,303lgKp
= –19,15TlgKp.
Поскольку
G
Н
Н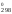 – ТS
– ТS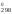 ,
справедливо выражение
,
справедливо выражение
–RTlnKp
Н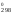 – ТS
– ТS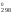 .
.
Используя уравнение изотермы для любой реакции при некоторых произвольно выбранных значениях давления и температуры можно рассчитать G (бесконечное множество значений) и сделать вывод о термодинамической вероятности протекания этой реакции при выбранных условиях.
Анализ многочисленных экспериментальных данных показал, что при реальных изменениях условий (парциальных давлений или концентраций реагирующих веществ) второе слагаемое в уравнении изотермы Вант-Гоффа не превышает 40 кДж (≤ 40 кДж). Это означает, что если /GоТ/ > 40 кДж, то вывод о возможности или невозможности протекания реакции в любых реальных условиях можно сделать просто по знаку GоТ.
Процесс
термодинамически невозможен
как самопроизвольный при G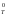 >> 0 (>40
кДж).
>> 0 (>40
кДж).
Если
G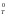 << 0 (<–40
кДж),
то процесс термодинамически возможен,
протекает в прямом направлении практически
необратимо.
<< 0 (<–40
кДж),
то процесс термодинамически возможен,
протекает в прямом направлении практически
необратимо.
Значения
G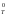 от
–40 кДж до +40 кДж
соответствуют обратимым
процессам.
от
–40 кДж до +40 кДж
соответствуют обратимым
процессам.
При
условии G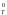 = 0 оба направления процессаравновероятны.
Температура,
при которой прямой и обратный процессы
равновероятны,
может быть определена в соответствии
с формулой (1):
= 0 оба направления процессаравновероятны.
Температура,
при которой прямой и обратный процессы
равновероятны,
может быть определена в соответствии
с формулой (1):
Т
=
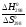 .
.
Вклады энтальпийного и энтропийного факторов существенно зависят от температуры.
Если Т → 0, то G
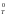 →
Н
→
Н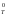 .
Таким образом, при низких температурах
величина и знак G
.
Таким образом, при низких температурах
величина и знак G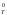 определяются величиной и знаком Н
определяются величиной и знаком Н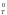 .
Принизких
температурах
самопроизвольно протекают, как правило,
экзотермические
реакции.
.
Принизких
температурах
самопроизвольно протекают, как правило,
экзотермические
реакции.Если Т → ∞, то G
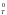 →
(–
Т
S
→
(–
Т
S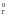 ).
При высоких температурах величина и
знак определяются величиной и знаком
S
).
При высоких температурах величина и
знак определяются величиной и знаком
S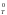 .
При высоких
температурах
самопроизвольно протекают, как правило,
реакции, ведущие к увеличению
энтропии.
.
При высоких
температурах
самопроизвольно протекают, как правило,
реакции, ведущие к увеличению
энтропии.
Таким
образом, в зависимости от температуры
влияние либо энтальпийного, либо
энтропийного факторов на значение и
знак G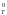 и,
следовательно, на направление может
быть определяющим.
и,
следовательно, на направление может
быть определяющим.
Табл. 1. - Влияние температуры на направление химических реакций
|
Н |
S |
G |
Принципиальная возможность и условия протекания реакции |
|
-- |
++ |
- |
Возможна при любой температуре |
|
›+ |
‹- |
- |
Принципиально невозможна. Возможна в обратном направлении |
|
-- |
-- |
± |
Возможна
при низких температурах (Т
<
|
|
++ |
++ |
± |
Возможна
при высоких температурах(Т
> |
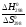 ).
).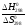 ).
).
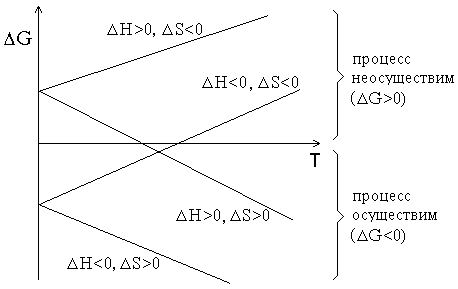
График зависимости G от температуры